DNA错配揭示了蛋白质-DNA识别中的构象缺陷
IF 50.5
1区 综合性期刊
Q1 MULTIDISCIPLINARY SCIENCES
引用次数: 51
摘要
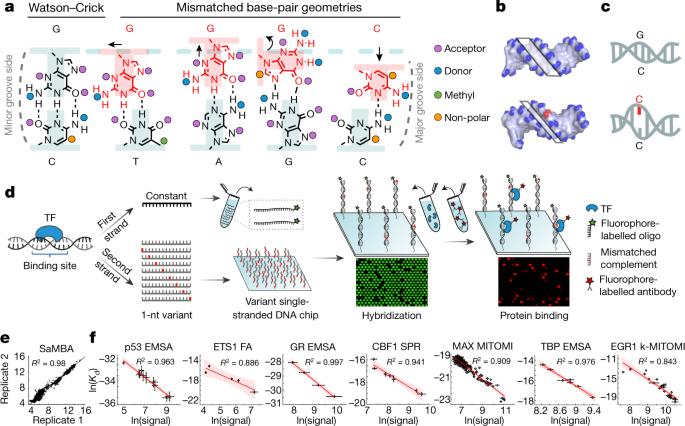
转录因子能识别特定的基因组序列,从而调控复杂的基因表达程序。尽管转录因子通过碱基读取和形状识别相结合的方式与特定 DNA 序列结合的事实已经得到证实,但人们对蛋白质与 DNA 结合的一些基本方面仍然知之甚少1,2。许多 DNA 结合蛋白会诱导 B-DNA 固有包膜之外的 DNA 结构发生变化。然而,由于扭曲的 DNA 在未结合的组合中存在的丰度很低3-9,因此与 DNA 扭曲相关的能量成本如何促进识别还很难研究。在这里,我们使用一种高通量检测方法--我们称之为 SaMBA(饱和错配结合检测)--来研究 DNA 构象惩罚在转录因子-DNA 识别中的作用。在 SaMBA 中,引入错配碱基对会预先引起 DNA 结构畸变,这种畸变比 Watson-Crick 序列变化引起的畸变要大得多。值得注意的是,大约 10%的错配会增加转录因子的结合力,在所研究的 22 个转录因子中,每个因子至少有一个错配会增加结合亲和力。错配还将非特异性位点转化为高亲和性位点,将高亲和性位点转化为 "超级位点",这些位点比任何已知的典型结合位点都表现出更强的亲和力。高分辨率 X 射线结构的测定以及核磁共振测量和结构分析表明,许多增加结合的 DNA 错配引起的扭曲与蛋白质结合引起的扭曲相似,从而预付了 DNA 变形所产生的部分能量成本。我们的工作表明,构象惩罚是蛋白质-DNA 识别的主要决定因素,并揭示了错配可招募转录因子从而调节细胞内复制和修复活动的机制10,11。一种在 DNA 序列中引入错配碱基对的高通量检测表明,错配可以通过预先支付 DNA 扭曲的部分能量成本来增加转录因子的结合亲和力。本文章由计算机程序翻译,如有差异,请以英文原文为准。
DNA mismatches reveal conformational penalties in protein–DNA recognition
Transcription factors recognize specific genomic sequences to regulate complex gene-expression programs. Although it is well-established that transcription factors bind to specific DNA sequences using a combination of base readout and shape recognition, some fundamental aspects of protein–DNA binding remain poorly understood1,2. Many DNA-binding proteins induce changes in the structure of the DNA outside the intrinsic B-DNA envelope. However, how the energetic cost that is associated with distorting the DNA contributes to recognition has proven difficult to study, because the distorted DNA exists in low abundance in the unbound ensemble3–9. Here we use a high-throughput assay that we term SaMBA (saturation mismatch-binding assay) to investigate the role of DNA conformational penalties in transcription factor–DNA recognition. In SaMBA, mismatched base pairs are introduced to pre-induce structural distortions in the DNA that are much larger than those induced by changes in the Watson–Crick sequence. Notably, approximately 10% of mismatches increased transcription factor binding, and for each of the 22 transcription factors that were examined, at least one mismatch was found that increased the binding affinity. Mismatches also converted non-specific sites into high-affinity sites, and high-affinity sites into ‘super sites’ that exhibit stronger affinity than any known canonical binding site. Determination of high-resolution X-ray structures, combined with nuclear magnetic resonance measurements and structural analyses, showed that many of the DNA mismatches that increase binding induce distortions that are similar to those induced by protein binding—thus prepaying some of the energetic cost incurred from deforming the DNA. Our work indicates that conformational penalties are a major determinant of protein–DNA recognition, and reveals mechanisms by which mismatches can recruit transcription factors and thus modulate replication and repair activities in the cell10,11. A high-throughput assay that introduces mismatched base pairs into the DNA sequence shows that mismatches can increase transcription factor binding affinity by prepaying some of the energetic cost of distorting the DNA.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature
综合性期刊-综合性期刊
CiteScore
90.00
自引率
1.20%
发文量
3652
审稿时长
3 months
期刊介绍:
Nature is a prestigious international journal that publishes peer-reviewed research in various scientific and technological fields. The selection of articles is based on criteria such as originality, importance, interdisciplinary relevance, timeliness, accessibility, elegance, and surprising conclusions. In addition to showcasing significant scientific advances, Nature delivers rapid, authoritative, insightful news, and interpretation of current and upcoming trends impacting science, scientists, and the broader public. The journal serves a dual purpose: firstly, to promptly share noteworthy scientific advances and foster discussions among scientists, and secondly, to ensure the swift dissemination of scientific results globally, emphasizing their significance for knowledge, culture, and daily life.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


