Tuqa Alkhateeb, Isatou Bah, Ajinkya Kumbhare, Dima Youssef, Zhi Q Yao, Charles E McCall, Mohamed El Gazzar
{"title":"长链非编码RNA Hotairm1促进S100A9在脓毒症期间支持MDSC扩增","authors":"Tuqa Alkhateeb, Isatou Bah, Ajinkya Kumbhare, Dima Youssef, Zhi Q Yao, Charles E McCall, Mohamed El Gazzar","doi":"","DOIUrl":null,"url":null,"abstract":"<p><p>Myeloid-derived suppressor cells (MDSCs) expand during mouse and human sepsis, but the mechanism responsible for this is unclear. We previously reported that nuclear transport of S100A9 protein programs Gr1<sup>+</sup>CD11b<sup>+</sup> myeloid precursors into MDSCs in septic mice. Here, we show that long non-coding RNA Hotairm1 converts MDSCs from an activator to a repressor state. Mechanistically, increased Hotairm1 expression in MDSCs in mice converted S100A9 from a secreted proinflammatory mediator to an immune repressor by binding to and shuttling it from cytosol to nucleus during late sepsis. High Hotairm1 levels were detected in exosomes shed from MDSCs from late septic mice. These exosomes inhibited lipopolysaccharide-stimulated secretion of S100A9 from early sepsis Gr1<sup>+</sup>CD11b<sup>+</sup> cells. Importantly, Hotairm1 knockdown in late sepsis Gr1<sup>+</sup>CD11b<sup>+</sup> MDSCs prevented S100A9 cytosol to nuclear transfer and decreased repression of proimmune T cells. Notably, ectopic expression of Hotairm1 in early sepsis Gr1<sup>+</sup>CD11b<sup>+</sup> cells shuttled S100A9 to the nucleus and promoted the MDSC repressor phenotype. In support of translating the mechanistic concept to human sepsis, we found that Hotairm1 binds S100A9 protein in CD33<sup>+</sup>CD11b<sup>+</sup>HLA-DR<sup>-</sup> MDSCs during established sepsis. Together, these data support that Hotairm1 is a plausible molecular target for treating late sepsis immune suppression in humans and its immune repressor mechanism may be cell autonomous.</p>","PeriodicalId":15473,"journal":{"name":"Journal of clinical & cellular immunology","volume":"11 6","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2020-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7744002/pdf/","citationCount":"0","resultStr":"{\"title\":\"Long Non-Coding RNA Hotairm1 Promotes S100A9 Support of MDSC Expansion during Sepsis.\",\"authors\":\"Tuqa Alkhateeb, Isatou Bah, Ajinkya Kumbhare, Dima Youssef, Zhi Q Yao, Charles E McCall, Mohamed El Gazzar\",\"doi\":\"\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Myeloid-derived suppressor cells (MDSCs) expand during mouse and human sepsis, but the mechanism responsible for this is unclear. We previously reported that nuclear transport of S100A9 protein programs Gr1<sup>+</sup>CD11b<sup>+</sup> myeloid precursors into MDSCs in septic mice. Here, we show that long non-coding RNA Hotairm1 converts MDSCs from an activator to a repressor state. Mechanistically, increased Hotairm1 expression in MDSCs in mice converted S100A9 from a secreted proinflammatory mediator to an immune repressor by binding to and shuttling it from cytosol to nucleus during late sepsis. High Hotairm1 levels were detected in exosomes shed from MDSCs from late septic mice. These exosomes inhibited lipopolysaccharide-stimulated secretion of S100A9 from early sepsis Gr1<sup>+</sup>CD11b<sup>+</sup> cells. Importantly, Hotairm1 knockdown in late sepsis Gr1<sup>+</sup>CD11b<sup>+</sup> MDSCs prevented S100A9 cytosol to nuclear transfer and decreased repression of proimmune T cells. Notably, ectopic expression of Hotairm1 in early sepsis Gr1<sup>+</sup>CD11b<sup>+</sup> cells shuttled S100A9 to the nucleus and promoted the MDSC repressor phenotype. In support of translating the mechanistic concept to human sepsis, we found that Hotairm1 binds S100A9 protein in CD33<sup>+</sup>CD11b<sup>+</sup>HLA-DR<sup>-</sup> MDSCs during established sepsis. Together, these data support that Hotairm1 is a plausible molecular target for treating late sepsis immune suppression in humans and its immune repressor mechanism may be cell autonomous.</p>\",\"PeriodicalId\":15473,\"journal\":{\"name\":\"Journal of clinical & cellular immunology\",\"volume\":\"11 6\",\"pages\":\"\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2020-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7744002/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of clinical & cellular immunology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2020/9/22 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of clinical & cellular immunology","FirstCategoryId":"1085","ListUrlMain":"","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2020/9/22 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Long Non-Coding RNA Hotairm1 Promotes S100A9 Support of MDSC Expansion during Sepsis.
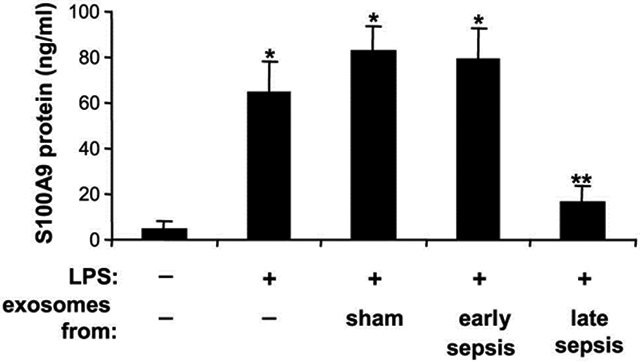
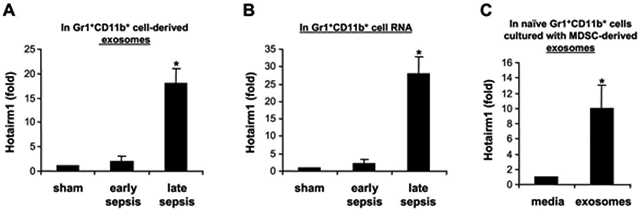
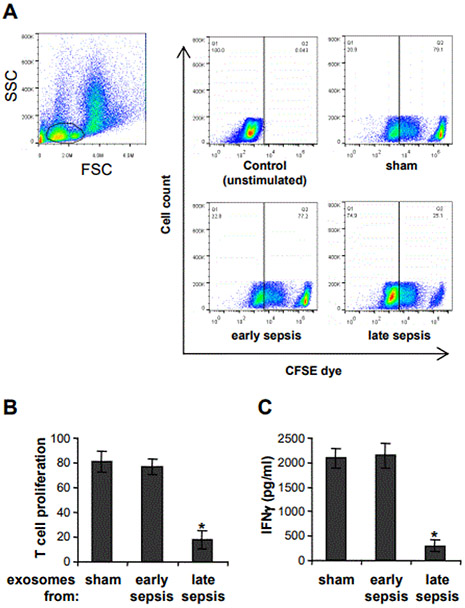
Myeloid-derived suppressor cells (MDSCs) expand during mouse and human sepsis, but the mechanism responsible for this is unclear. We previously reported that nuclear transport of S100A9 protein programs Gr1+CD11b+ myeloid precursors into MDSCs in septic mice. Here, we show that long non-coding RNA Hotairm1 converts MDSCs from an activator to a repressor state. Mechanistically, increased Hotairm1 expression in MDSCs in mice converted S100A9 from a secreted proinflammatory mediator to an immune repressor by binding to and shuttling it from cytosol to nucleus during late sepsis. High Hotairm1 levels were detected in exosomes shed from MDSCs from late septic mice. These exosomes inhibited lipopolysaccharide-stimulated secretion of S100A9 from early sepsis Gr1+CD11b+ cells. Importantly, Hotairm1 knockdown in late sepsis Gr1+CD11b+ MDSCs prevented S100A9 cytosol to nuclear transfer and decreased repression of proimmune T cells. Notably, ectopic expression of Hotairm1 in early sepsis Gr1+CD11b+ cells shuttled S100A9 to the nucleus and promoted the MDSC repressor phenotype. In support of translating the mechanistic concept to human sepsis, we found that Hotairm1 binds S100A9 protein in CD33+CD11b+HLA-DR- MDSCs during established sepsis. Together, these data support that Hotairm1 is a plausible molecular target for treating late sepsis immune suppression in humans and its immune repressor mechanism may be cell autonomous.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


