{"title":"InteractionGraphNet:一种新颖高效的深度图表示学习框架,用于准确预测蛋白质-配体相互作用","authors":"Dejun Jiang, Chang-Yu Hsieh, Zhenxing Wu, Yu Kang, Jike Wang, Ercheng Wang, Ben Liao, Chao Shen, Lei Xu, Jian Wu*, Dongsheng Cao*, Tingjun Hou*","doi":"10.1021/acs.jmedchem.1c01830","DOIUrl":null,"url":null,"abstract":"<p >Accurate quantification of protein–ligand interactions remains a key challenge to structure-based drug design. However, traditional machine learning (ML)-based methods based on handcrafted descriptors, one-dimensional protein sequences, and/or two-dimensional graph representations limit their capability to learn the generalized molecular interactions in 3D space. Here, we proposed a novel deep graph representation learning framework named InteractionGraphNet (IGN) to learn the protein–ligand interactions from the 3D structures of protein–ligand complexes. In IGN, two independent graph convolution modules were stacked to sequentially learn the intramolecular and intermolecular interactions, and the learned intermolecular interactions can be efficiently used for subsequent tasks. Extensive binding affinity prediction, large-scale structure-based virtual screening, and pose prediction experiments demonstrated that IGN achieved better or competitive performance against other state-of-the-art ML-based baselines and docking programs. More importantly, such state-of-the-art performance was proven from the successful learning of the key features in protein–ligand interactions instead of just memorizing certain biased patterns from data.</p>","PeriodicalId":46,"journal":{"name":"Journal of Medicinal Chemistry","volume":"64 24","pages":"18209–18232"},"PeriodicalIF":6.8000,"publicationDate":"2021-12-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"74","resultStr":"{\"title\":\"InteractionGraphNet: A Novel and Efficient Deep Graph Representation Learning Framework for Accurate Protein–Ligand Interaction Predictions\",\"authors\":\"Dejun Jiang, Chang-Yu Hsieh, Zhenxing Wu, Yu Kang, Jike Wang, Ercheng Wang, Ben Liao, Chao Shen, Lei Xu, Jian Wu*, Dongsheng Cao*, Tingjun Hou*\",\"doi\":\"10.1021/acs.jmedchem.1c01830\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Accurate quantification of protein–ligand interactions remains a key challenge to structure-based drug design. However, traditional machine learning (ML)-based methods based on handcrafted descriptors, one-dimensional protein sequences, and/or two-dimensional graph representations limit their capability to learn the generalized molecular interactions in 3D space. Here, we proposed a novel deep graph representation learning framework named InteractionGraphNet (IGN) to learn the protein–ligand interactions from the 3D structures of protein–ligand complexes. In IGN, two independent graph convolution modules were stacked to sequentially learn the intramolecular and intermolecular interactions, and the learned intermolecular interactions can be efficiently used for subsequent tasks. Extensive binding affinity prediction, large-scale structure-based virtual screening, and pose prediction experiments demonstrated that IGN achieved better or competitive performance against other state-of-the-art ML-based baselines and docking programs. More importantly, such state-of-the-art performance was proven from the successful learning of the key features in protein–ligand interactions instead of just memorizing certain biased patterns from data.</p>\",\"PeriodicalId\":46,\"journal\":{\"name\":\"Journal of Medicinal Chemistry\",\"volume\":\"64 24\",\"pages\":\"18209–18232\"},\"PeriodicalIF\":6.8000,\"publicationDate\":\"2021-12-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"74\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Medicinal Chemistry\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c01830\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c01830","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
InteractionGraphNet: A Novel and Efficient Deep Graph Representation Learning Framework for Accurate Protein–Ligand Interaction Predictions
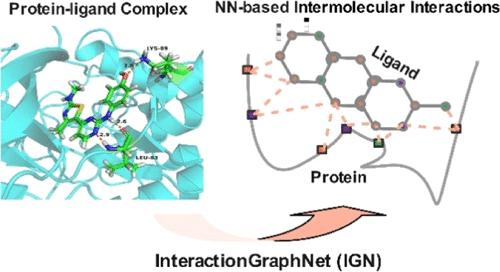
Accurate quantification of protein–ligand interactions remains a key challenge to structure-based drug design. However, traditional machine learning (ML)-based methods based on handcrafted descriptors, one-dimensional protein sequences, and/or two-dimensional graph representations limit their capability to learn the generalized molecular interactions in 3D space. Here, we proposed a novel deep graph representation learning framework named InteractionGraphNet (IGN) to learn the protein–ligand interactions from the 3D structures of protein–ligand complexes. In IGN, two independent graph convolution modules were stacked to sequentially learn the intramolecular and intermolecular interactions, and the learned intermolecular interactions can be efficiently used for subsequent tasks. Extensive binding affinity prediction, large-scale structure-based virtual screening, and pose prediction experiments demonstrated that IGN achieved better or competitive performance against other state-of-the-art ML-based baselines and docking programs. More importantly, such state-of-the-art performance was proven from the successful learning of the key features in protein–ligand interactions instead of just memorizing certain biased patterns from data.
期刊介绍:
The Journal of Medicinal Chemistry is a prestigious biweekly peer-reviewed publication that focuses on the multifaceted field of medicinal chemistry. Since its inception in 1959 as the Journal of Medicinal and Pharmaceutical Chemistry, it has evolved to become a cornerstone in the dissemination of research findings related to the design, synthesis, and development of therapeutic agents.
The Journal of Medicinal Chemistry is recognized for its significant impact in the scientific community, as evidenced by its 2022 impact factor of 7.3. This metric reflects the journal's influence and the importance of its content in shaping the future of drug discovery and development. The journal serves as a vital resource for chemists, pharmacologists, and other researchers interested in the molecular mechanisms of drug action and the optimization of therapeutic compounds.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


