Jian Gao, Li Liang, Yasheng Zhu, Shengzhi Qiu, Tao Wang, Ling Zhang
{"title":"基于配体和结构的多肽去甲酰基酶抑制剂抗菌药物鉴定方法。","authors":"Jian Gao, Li Liang, Yasheng Zhu, Shengzhi Qiu, Tao Wang, Ling Zhang","doi":"10.3390/ijms17071141","DOIUrl":null,"url":null,"abstract":"<p><p>Peptide deformylase (PDF) is a metalloprotease catalyzing the removal of a formyl group from newly synthesized proteins, which makes it an important antibacterial drug target. Given the importance of PDF inhibitors like actinonin in antibacterial drug discovery, several reported potent PDF inhibitors were used to develop pharmacophore models using the Galahad module of Sybyl 7.1 software. Generated pharmacophore models were composed of two donor atom centers, four acceptor atom centers and two hydrophobic groups. Model-1 was screened against the Zinc database and several compounds were retrieved as hits. Compounds with Qfit values of more than 60 were employed to perform a molecular docking study with the receptor Escherichia coli PDF, then compounds with docking score values of more than 6 were used to predict the in silico pharmacokinetic and toxicity risk via OSIRIS property explorer. Two known PDF inhibitors were also used to perform a molecular docking study with E. coli PDF as reference molecules. The results of the molecular docking study were validated by reproducing the crystal structure of actinonin. Molecular docking and in silico pharmacokinetic and toxicity prediction studies suggested that ZINC08740166 has a relatively high docking score of 7.44 and a drug score of 0.78. </p>","PeriodicalId":14156,"journal":{"name":"International Journal of Molecular Sciences","volume":"17 7","pages":""},"PeriodicalIF":5.6000,"publicationDate":"2016-07-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.3390/ijms17071141","citationCount":"26","resultStr":"{\"title\":\"Ligand and Structure-Based Approaches for the Identification of Peptide Deformylase Inhibitors as Antibacterial Drugs.\",\"authors\":\"Jian Gao, Li Liang, Yasheng Zhu, Shengzhi Qiu, Tao Wang, Ling Zhang\",\"doi\":\"10.3390/ijms17071141\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Peptide deformylase (PDF) is a metalloprotease catalyzing the removal of a formyl group from newly synthesized proteins, which makes it an important antibacterial drug target. Given the importance of PDF inhibitors like actinonin in antibacterial drug discovery, several reported potent PDF inhibitors were used to develop pharmacophore models using the Galahad module of Sybyl 7.1 software. Generated pharmacophore models were composed of two donor atom centers, four acceptor atom centers and two hydrophobic groups. Model-1 was screened against the Zinc database and several compounds were retrieved as hits. Compounds with Qfit values of more than 60 were employed to perform a molecular docking study with the receptor Escherichia coli PDF, then compounds with docking score values of more than 6 were used to predict the in silico pharmacokinetic and toxicity risk via OSIRIS property explorer. Two known PDF inhibitors were also used to perform a molecular docking study with E. coli PDF as reference molecules. The results of the molecular docking study were validated by reproducing the crystal structure of actinonin. Molecular docking and in silico pharmacokinetic and toxicity prediction studies suggested that ZINC08740166 has a relatively high docking score of 7.44 and a drug score of 0.78. </p>\",\"PeriodicalId\":14156,\"journal\":{\"name\":\"International Journal of Molecular Sciences\",\"volume\":\"17 7\",\"pages\":\"\"},\"PeriodicalIF\":5.6000,\"publicationDate\":\"2016-07-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.3390/ijms17071141\",\"citationCount\":\"26\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Molecular Sciences\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.3390/ijms17071141\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Molecular Sciences","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.3390/ijms17071141","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Ligand and Structure-Based Approaches for the Identification of Peptide Deformylase Inhibitors as Antibacterial Drugs.
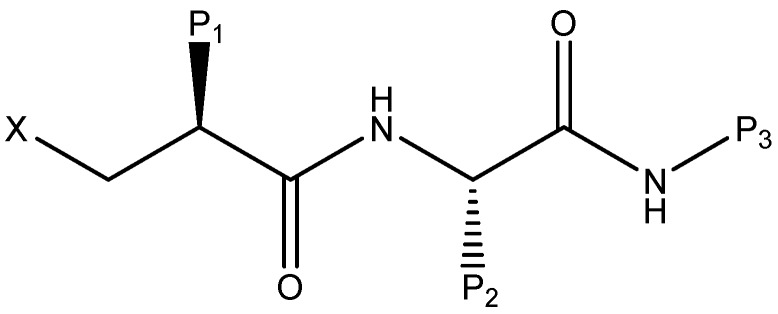
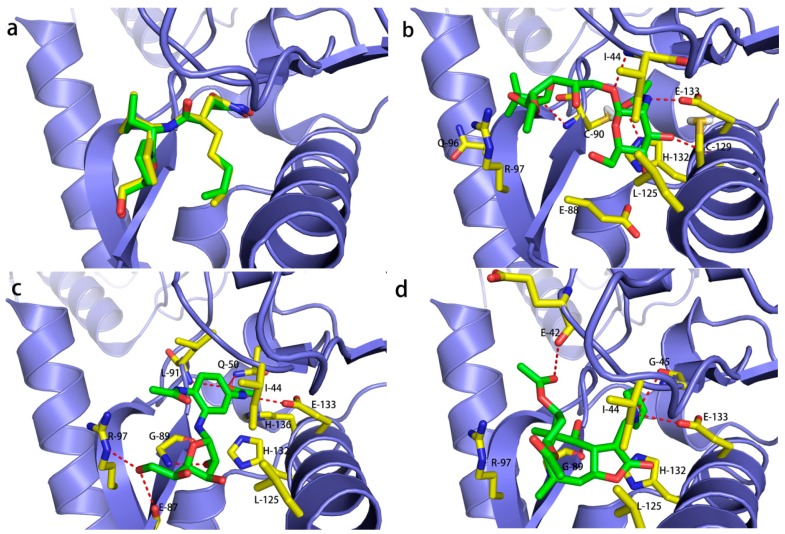
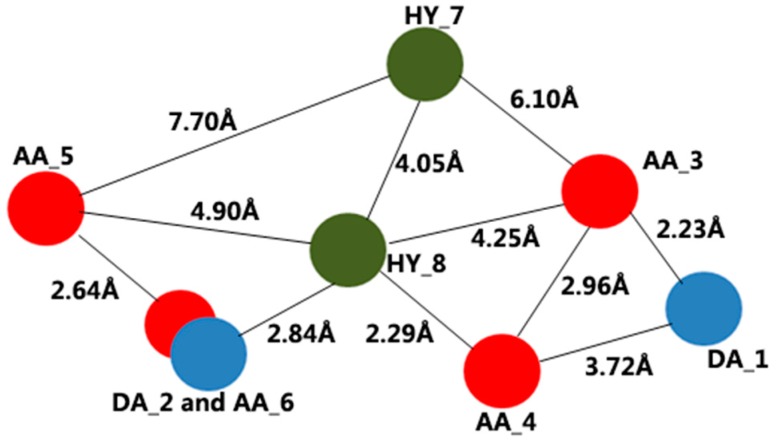
Peptide deformylase (PDF) is a metalloprotease catalyzing the removal of a formyl group from newly synthesized proteins, which makes it an important antibacterial drug target. Given the importance of PDF inhibitors like actinonin in antibacterial drug discovery, several reported potent PDF inhibitors were used to develop pharmacophore models using the Galahad module of Sybyl 7.1 software. Generated pharmacophore models were composed of two donor atom centers, four acceptor atom centers and two hydrophobic groups. Model-1 was screened against the Zinc database and several compounds were retrieved as hits. Compounds with Qfit values of more than 60 were employed to perform a molecular docking study with the receptor Escherichia coli PDF, then compounds with docking score values of more than 6 were used to predict the in silico pharmacokinetic and toxicity risk via OSIRIS property explorer. Two known PDF inhibitors were also used to perform a molecular docking study with E. coli PDF as reference molecules. The results of the molecular docking study were validated by reproducing the crystal structure of actinonin. Molecular docking and in silico pharmacokinetic and toxicity prediction studies suggested that ZINC08740166 has a relatively high docking score of 7.44 and a drug score of 0.78.
期刊介绍:
The International Journal of Molecular Sciences (ISSN 1422-0067) provides an advanced forum for chemistry, molecular physics (chemical physics and physical chemistry) and molecular biology. It publishes research articles, reviews, communications and short notes. Our aim is to encourage scientists to publish their theoretical and experimental results in as much detail as possible. Therefore, there is no restriction on the length of the papers or the number of electronics supplementary files. For articles with computational results, the full experimental details must be provided so that the results can be reproduced. Electronic files regarding the full details of the calculation and experimental procedure, if unable to be published in a normal way, can be deposited as supplementary material (including animated pictures, videos, interactive Excel sheets, software executables and others).

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


