William H Bradley, Kevin Eng, Min Le, A Craig Mackinnon, Christina Kendziorski, Janet S Rader
{"title":"比较福尔马林固定组织、石蜡包埋组织和qPCR的基因表达数据与速冻组织和微阵列的基因表达数据,以模拟卵巢癌患者的预后。","authors":"William H Bradley, Kevin Eng, Min Le, A Craig Mackinnon, Christina Kendziorski, Janet S Rader","doi":"10.1186/s12907-015-0017-1","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Previously, we have used clinical and gene expression data from The Cancer Genome Atlas (TCGA) to model a pathway-based index predicting outcomes in ovarian carcinoma. This data were obtained from snap-frozen tissue measured with the Affymetrix U133 platform. In the current study, we correlate the data used to model with data derived from TaqMan qPCR both snap frozen and paraffin embedded (FFPE) samples.</p><p><strong>Methods: </strong>To compare the effect of preservation methods on gene expression measured by qPCR, we assessed 18 patient and tumor sample matched snap-frozen and FFPE ovarian carcinoma samples. To compare gene measurement technologies, we correlated qPCR data from 10 patients with tumor sample matched snap-frozen ovarian carcinoma samples with the microarray data from TCGA. We normalized results to the average expression of three housekeeping genes. We scaled and centered the data for comparison to the Affymetrix output.</p><p><strong>Results: </strong>For the 18 specimens, gene expression data obtained from snap-frozen tissue correlated highly with that from FFPE samples in our TaqMan assay (r > 0.82). For the 10 duplicate TCGA specimens, the reported microarray data correlated well (r = 0.6) with our qPCR data, and ranges of expression along pathways were similar.</p><p><strong>Conclusions: </strong>Gene expression data obtained by qPCR from FFPE serous ovarian carcinoma samples can be used to assess in the pathway-based predictive model. The normalization procedures described control variations in expression, and the range calculated along a specific pathway can be interpreted for a patient's risk profile.</p>","PeriodicalId":35804,"journal":{"name":"BMC Clinical Pathology","volume":"15 ","pages":"17"},"PeriodicalIF":0.0000,"publicationDate":"2015-09-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/s12907-015-0017-1","citationCount":"13","resultStr":"{\"title\":\"Comparing gene expression data from formalin-fixed, paraffin embedded tissues and qPCR with that from snap-frozen tissue and microarrays for modeling outcomes of patients with ovarian carcinoma.\",\"authors\":\"William H Bradley, Kevin Eng, Min Le, A Craig Mackinnon, Christina Kendziorski, Janet S Rader\",\"doi\":\"10.1186/s12907-015-0017-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Previously, we have used clinical and gene expression data from The Cancer Genome Atlas (TCGA) to model a pathway-based index predicting outcomes in ovarian carcinoma. This data were obtained from snap-frozen tissue measured with the Affymetrix U133 platform. In the current study, we correlate the data used to model with data derived from TaqMan qPCR both snap frozen and paraffin embedded (FFPE) samples.</p><p><strong>Methods: </strong>To compare the effect of preservation methods on gene expression measured by qPCR, we assessed 18 patient and tumor sample matched snap-frozen and FFPE ovarian carcinoma samples. To compare gene measurement technologies, we correlated qPCR data from 10 patients with tumor sample matched snap-frozen ovarian carcinoma samples with the microarray data from TCGA. We normalized results to the average expression of three housekeeping genes. We scaled and centered the data for comparison to the Affymetrix output.</p><p><strong>Results: </strong>For the 18 specimens, gene expression data obtained from snap-frozen tissue correlated highly with that from FFPE samples in our TaqMan assay (r > 0.82). For the 10 duplicate TCGA specimens, the reported microarray data correlated well (r = 0.6) with our qPCR data, and ranges of expression along pathways were similar.</p><p><strong>Conclusions: </strong>Gene expression data obtained by qPCR from FFPE serous ovarian carcinoma samples can be used to assess in the pathway-based predictive model. The normalization procedures described control variations in expression, and the range calculated along a specific pathway can be interpreted for a patient's risk profile.</p>\",\"PeriodicalId\":35804,\"journal\":{\"name\":\"BMC Clinical Pathology\",\"volume\":\"15 \",\"pages\":\"17\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2015-09-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/s12907-015-0017-1\",\"citationCount\":\"13\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Clinical Pathology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s12907-015-0017-1\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2015/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Clinical Pathology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s12907-015-0017-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2015/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"Medicine","Score":null,"Total":0}
Comparing gene expression data from formalin-fixed, paraffin embedded tissues and qPCR with that from snap-frozen tissue and microarrays for modeling outcomes of patients with ovarian carcinoma.
Background: Previously, we have used clinical and gene expression data from The Cancer Genome Atlas (TCGA) to model a pathway-based index predicting outcomes in ovarian carcinoma. This data were obtained from snap-frozen tissue measured with the Affymetrix U133 platform. In the current study, we correlate the data used to model with data derived from TaqMan qPCR both snap frozen and paraffin embedded (FFPE) samples.
Methods: To compare the effect of preservation methods on gene expression measured by qPCR, we assessed 18 patient and tumor sample matched snap-frozen and FFPE ovarian carcinoma samples. To compare gene measurement technologies, we correlated qPCR data from 10 patients with tumor sample matched snap-frozen ovarian carcinoma samples with the microarray data from TCGA. We normalized results to the average expression of three housekeeping genes. We scaled and centered the data for comparison to the Affymetrix output.
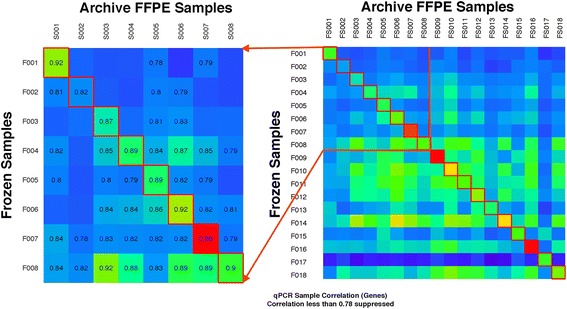
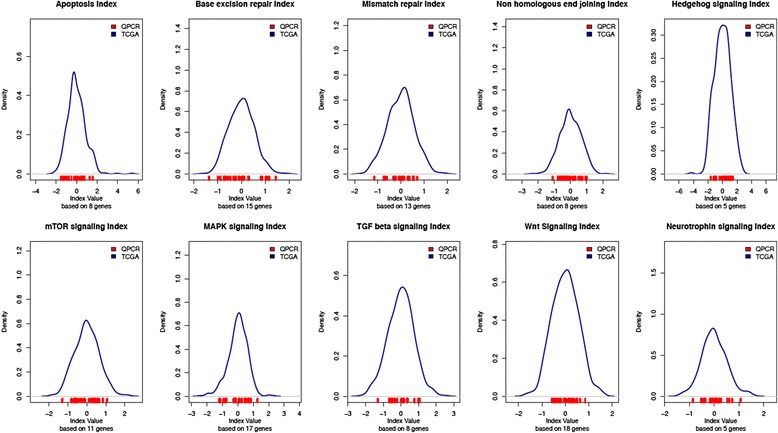
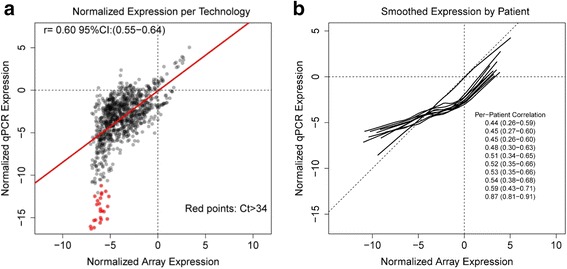
Results: For the 18 specimens, gene expression data obtained from snap-frozen tissue correlated highly with that from FFPE samples in our TaqMan assay (r > 0.82). For the 10 duplicate TCGA specimens, the reported microarray data correlated well (r = 0.6) with our qPCR data, and ranges of expression along pathways were similar.
Conclusions: Gene expression data obtained by qPCR from FFPE serous ovarian carcinoma samples can be used to assess in the pathway-based predictive model. The normalization procedures described control variations in expression, and the range calculated along a specific pathway can be interpreted for a patient's risk profile.
期刊介绍:
BMC Clinical Pathology is an open access journal publishing original peer-reviewed research articles in all aspects of histopathology, haematology, clinical biochemistry, and medical microbiology (including virology, parasitology, and infection control). BMC Clinical Pathology (ISSN 1472-6890) is indexed/tracked/covered by PubMed, CAS, EMBASE, Scopus and Google Scholar.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


