Guillermo Gervasini, Maria J Caballero, Juan A Carrillo, Julio Benitez
{"title":"非典型抗精神病药物对细胞色素p450体外抑制作用的比较。","authors":"Guillermo Gervasini, Maria J Caballero, Juan A Carrillo, Julio Benitez","doi":"10.1155/2013/792456","DOIUrl":null,"url":null,"abstract":"<p><p>The goal of this study was to assess in human liver microsomes the inhibitory capacity of commonly used antipsychotics on the most prominent CYP450 drug metabolizing enzymes (CYP1A2, CYP2C9, CYP2D6, and CYP3A). Chlorpromazine was the only antipsychotic that inhibited CYP1A2 activity (IC50 = 9.5 μ M), whilst levomepromazine, chlorpromazine, and thioridazine significantly decreased CYP2D6-mediated formation of 1'-hydroxybufuralol (IC50 range, 3.5-25.5 μ M). Olanzapine inhibited CYP3A-catalyzed production of 1', and 4'-hydroxymidazolam (IC50 = 14.65 and 42.20 μ M, resp.). In contrast, risperidone (IC50 = 20.7 μ M) and levomepromazine (IC50 = 30 μ M) showed selectivity towards the inhibition of midazolam 1'-hydroxylation reaction, and haloperidol did so towards 4'-hydroxylation (IC50 of 2.76 μ M). Thioridazine displayed a Ki of 1.75 μ M and an inhibitory potency of 1.57 on CYP2D6, suggesting a potential to induce in vivo interactions. However, with this exception, and given the observed Ki values, the potential of the assayed antipsychotics to produce clinically significant inhibitions of CYP450 isoforms in vivo seems limited.</p>","PeriodicalId":14662,"journal":{"name":"ISRN Pharmacology","volume":"2013 ","pages":"792456"},"PeriodicalIF":0.0000,"publicationDate":"2013-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1155/2013/792456","citationCount":"15","resultStr":"{\"title\":\"Comparative cytochrome p450 in vitro inhibition by atypical antipsychotic drugs.\",\"authors\":\"Guillermo Gervasini, Maria J Caballero, Juan A Carrillo, Julio Benitez\",\"doi\":\"10.1155/2013/792456\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The goal of this study was to assess in human liver microsomes the inhibitory capacity of commonly used antipsychotics on the most prominent CYP450 drug metabolizing enzymes (CYP1A2, CYP2C9, CYP2D6, and CYP3A). Chlorpromazine was the only antipsychotic that inhibited CYP1A2 activity (IC50 = 9.5 μ M), whilst levomepromazine, chlorpromazine, and thioridazine significantly decreased CYP2D6-mediated formation of 1'-hydroxybufuralol (IC50 range, 3.5-25.5 μ M). Olanzapine inhibited CYP3A-catalyzed production of 1', and 4'-hydroxymidazolam (IC50 = 14.65 and 42.20 μ M, resp.). In contrast, risperidone (IC50 = 20.7 μ M) and levomepromazine (IC50 = 30 μ M) showed selectivity towards the inhibition of midazolam 1'-hydroxylation reaction, and haloperidol did so towards 4'-hydroxylation (IC50 of 2.76 μ M). Thioridazine displayed a Ki of 1.75 μ M and an inhibitory potency of 1.57 on CYP2D6, suggesting a potential to induce in vivo interactions. However, with this exception, and given the observed Ki values, the potential of the assayed antipsychotics to produce clinically significant inhibitions of CYP450 isoforms in vivo seems limited.</p>\",\"PeriodicalId\":14662,\"journal\":{\"name\":\"ISRN Pharmacology\",\"volume\":\"2013 \",\"pages\":\"792456\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2013-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1155/2013/792456\",\"citationCount\":\"15\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ISRN Pharmacology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/2013/792456\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2013/2/13 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ISRN Pharmacology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/2013/792456","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2013/2/13 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Comparative cytochrome p450 in vitro inhibition by atypical antipsychotic drugs.
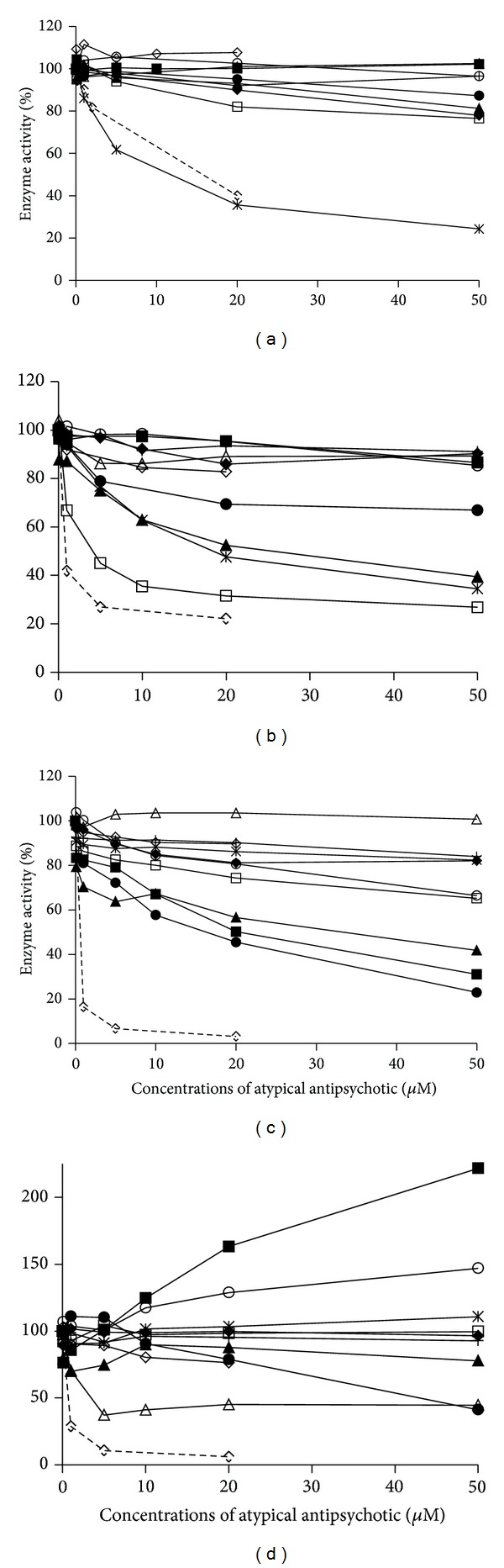
The goal of this study was to assess in human liver microsomes the inhibitory capacity of commonly used antipsychotics on the most prominent CYP450 drug metabolizing enzymes (CYP1A2, CYP2C9, CYP2D6, and CYP3A). Chlorpromazine was the only antipsychotic that inhibited CYP1A2 activity (IC50 = 9.5 μ M), whilst levomepromazine, chlorpromazine, and thioridazine significantly decreased CYP2D6-mediated formation of 1'-hydroxybufuralol (IC50 range, 3.5-25.5 μ M). Olanzapine inhibited CYP3A-catalyzed production of 1', and 4'-hydroxymidazolam (IC50 = 14.65 and 42.20 μ M, resp.). In contrast, risperidone (IC50 = 20.7 μ M) and levomepromazine (IC50 = 30 μ M) showed selectivity towards the inhibition of midazolam 1'-hydroxylation reaction, and haloperidol did so towards 4'-hydroxylation (IC50 of 2.76 μ M). Thioridazine displayed a Ki of 1.75 μ M and an inhibitory potency of 1.57 on CYP2D6, suggesting a potential to induce in vivo interactions. However, with this exception, and given the observed Ki values, the potential of the assayed antipsychotics to produce clinically significant inhibitions of CYP450 isoforms in vivo seems limited.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


