{"title":"从瘢痕疙瘩成纤维细胞基因网络的逆向工程中获得的见解。","authors":"Brandon N S Ooi, Toan Thang Phan","doi":"10.1186/1742-4682-8-13","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Keloids are protrusive claw-like scars that have a propensity to recur even after surgery, and its molecular etiology remains elusive. The goal of reverse engineering is to infer gene networks from observational data, thus providing insight into the inner workings of a cell. However, most attempts at modeling biological networks have been done using simulated data. This study aims to highlight some of the issues involved in working with experimental data, and at the same time gain some insights into the transcriptional regulatory mechanism present in keloid fibroblasts.</p><p><strong>Methods: </strong>Microarray data from our previous study was combined with microarray data obtained from the literature as well as new microarray data generated by our group. For the physical approach, we used the fREDUCE algorithm for correlating expression values to binding motifs. For the influence approach, we compared the Bayesian algorithm BANJO with the information theoretic method ARACNE in terms of performance in recovering known influence networks obtained from the KEGG database. In addition, we also compared the performance of different normalization methods as well as different types of gene networks.</p><p><strong>Results: </strong>Using the physical approach, we found consensus sequences that were active in the keloid condition, as well as some sequences that were responsive to steroids, a commonly used treatment for keloids. From the influence approach, we found that BANJO was better at recovering the gene networks compared to ARACNE and that transcriptional networks were better suited for network recovery compared to cytokine-receptor interaction networks and intracellular signaling networks. We also found that the NFKB transcriptional network that was inferred from normal fibroblast data was more accurate compared to that inferred from keloid data, suggesting a more robust network in the keloid condition.</p><p><strong>Conclusions: </strong>Consensus sequences that were found from this study are possible transcription factor binding sites and could be explored for developing future keloid treatments or for improving the efficacy of current steroid treatments. We also found that the combination of the Bayesian algorithm, RMA normalization and transcriptional networks gave the best reconstruction results and this could serve as a guide for future influence approaches dealing with experimental data.</p>","PeriodicalId":51195,"journal":{"name":"Theoretical Biology and Medical Modelling","volume":" ","pages":"13"},"PeriodicalIF":0.0000,"publicationDate":"2011-05-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://sci-hub-pdf.com/10.1186/1742-4682-8-13","citationCount":"11","resultStr":"{\"title\":\"Insights gained from the reverse engineering of gene networks in keloid fibroblasts.\",\"authors\":\"Brandon N S Ooi, Toan Thang Phan\",\"doi\":\"10.1186/1742-4682-8-13\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Keloids are protrusive claw-like scars that have a propensity to recur even after surgery, and its molecular etiology remains elusive. The goal of reverse engineering is to infer gene networks from observational data, thus providing insight into the inner workings of a cell. However, most attempts at modeling biological networks have been done using simulated data. This study aims to highlight some of the issues involved in working with experimental data, and at the same time gain some insights into the transcriptional regulatory mechanism present in keloid fibroblasts.</p><p><strong>Methods: </strong>Microarray data from our previous study was combined with microarray data obtained from the literature as well as new microarray data generated by our group. For the physical approach, we used the fREDUCE algorithm for correlating expression values to binding motifs. For the influence approach, we compared the Bayesian algorithm BANJO with the information theoretic method ARACNE in terms of performance in recovering known influence networks obtained from the KEGG database. In addition, we also compared the performance of different normalization methods as well as different types of gene networks.</p><p><strong>Results: </strong>Using the physical approach, we found consensus sequences that were active in the keloid condition, as well as some sequences that were responsive to steroids, a commonly used treatment for keloids. From the influence approach, we found that BANJO was better at recovering the gene networks compared to ARACNE and that transcriptional networks were better suited for network recovery compared to cytokine-receptor interaction networks and intracellular signaling networks. We also found that the NFKB transcriptional network that was inferred from normal fibroblast data was more accurate compared to that inferred from keloid data, suggesting a more robust network in the keloid condition.</p><p><strong>Conclusions: </strong>Consensus sequences that were found from this study are possible transcription factor binding sites and could be explored for developing future keloid treatments or for improving the efficacy of current steroid treatments. We also found that the combination of the Bayesian algorithm, RMA normalization and transcriptional networks gave the best reconstruction results and this could serve as a guide for future influence approaches dealing with experimental data.</p>\",\"PeriodicalId\":51195,\"journal\":{\"name\":\"Theoretical Biology and Medical Modelling\",\"volume\":\" \",\"pages\":\"13\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2011-05-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://sci-hub-pdf.com/10.1186/1742-4682-8-13\",\"citationCount\":\"11\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Theoretical Biology and Medical Modelling\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/1742-4682-8-13\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"Mathematics\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Theoretical Biology and Medical Modelling","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/1742-4682-8-13","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Mathematics","Score":null,"Total":0}
Insights gained from the reverse engineering of gene networks in keloid fibroblasts.
Background: Keloids are protrusive claw-like scars that have a propensity to recur even after surgery, and its molecular etiology remains elusive. The goal of reverse engineering is to infer gene networks from observational data, thus providing insight into the inner workings of a cell. However, most attempts at modeling biological networks have been done using simulated data. This study aims to highlight some of the issues involved in working with experimental data, and at the same time gain some insights into the transcriptional regulatory mechanism present in keloid fibroblasts.
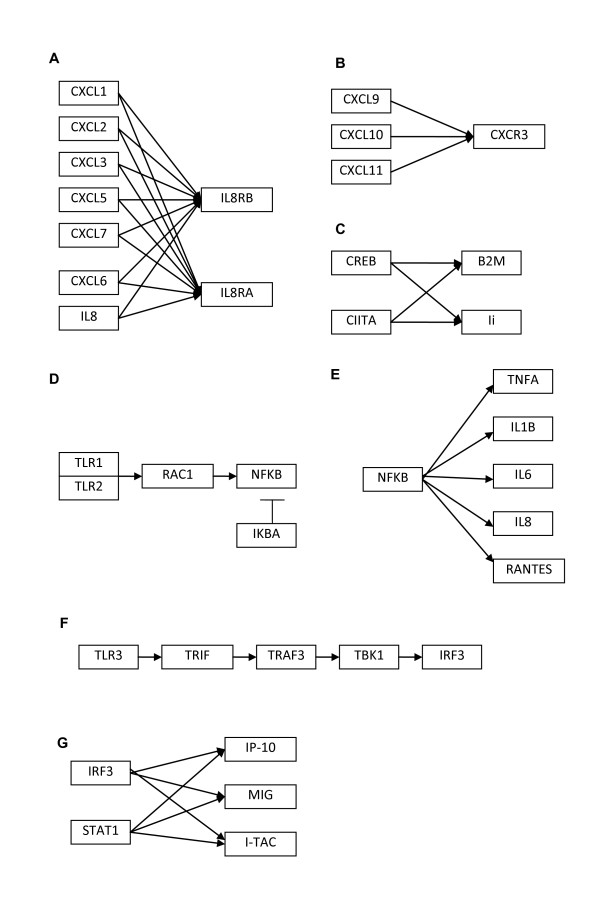
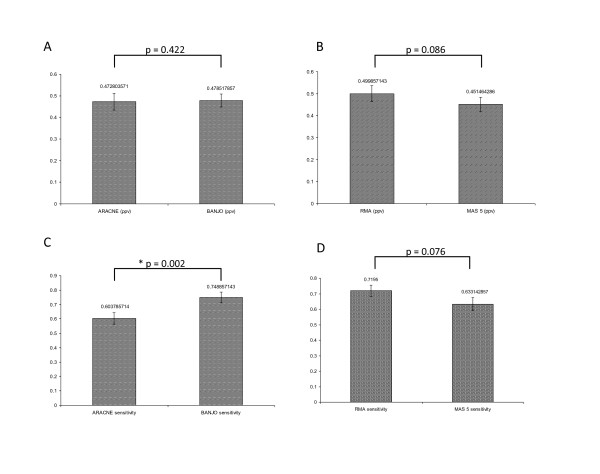
Methods: Microarray data from our previous study was combined with microarray data obtained from the literature as well as new microarray data generated by our group. For the physical approach, we used the fREDUCE algorithm for correlating expression values to binding motifs. For the influence approach, we compared the Bayesian algorithm BANJO with the information theoretic method ARACNE in terms of performance in recovering known influence networks obtained from the KEGG database. In addition, we also compared the performance of different normalization methods as well as different types of gene networks.
Results: Using the physical approach, we found consensus sequences that were active in the keloid condition, as well as some sequences that were responsive to steroids, a commonly used treatment for keloids. From the influence approach, we found that BANJO was better at recovering the gene networks compared to ARACNE and that transcriptional networks were better suited for network recovery compared to cytokine-receptor interaction networks and intracellular signaling networks. We also found that the NFKB transcriptional network that was inferred from normal fibroblast data was more accurate compared to that inferred from keloid data, suggesting a more robust network in the keloid condition.
Conclusions: Consensus sequences that were found from this study are possible transcription factor binding sites and could be explored for developing future keloid treatments or for improving the efficacy of current steroid treatments. We also found that the combination of the Bayesian algorithm, RMA normalization and transcriptional networks gave the best reconstruction results and this could serve as a guide for future influence approaches dealing with experimental data.
期刊介绍:
Theoretical Biology and Medical Modelling is an open access peer-reviewed journal adopting a broad definition of "biology" and focusing on theoretical ideas and models associated with developments in biology and medicine. Mathematicians, biologists and clinicians of various specialisms, philosophers and historians of science are all contributing to the emergence of novel concepts in an age of systems biology, bioinformatics and computer modelling. This is the field in which Theoretical Biology and Medical Modelling operates. We welcome submissions that are technically sound and offering either improved understanding in biology and medicine or progress in theory or method.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


