Martí López , Kai S. Exner , Francesc Viñes , Francesc Illas
{"title":"V2C MXene析氢反应机理的理论研究:热力学和动力学方面","authors":"Martí López , Kai S. Exner , Francesc Viñes , Francesc Illas","doi":"10.1016/j.jcat.2023.03.027","DOIUrl":null,"url":null,"abstract":"<div><p>Both experimentally and theoretically, the MXene family has shown promising hydrogen evolution reaction (HER) capabilities. However, so far, the theoretical approach has been relying on the well-known thermodynamic descriptor, Δ<em>G</em><sub>H</sub>, whereas experimental studies report Tafel plots, containing kinetic rather than thermodynamic information. Aiming to link theory to experiments, the present study explores five different HER pathways over the exemplary V<sub>2</sub>C (0001) MXene by density functional theory calculations. While the surface coverage under HER conditions (with either H* or OH* adsorbates) is extracted from a Pourbaix diagram, we determine the energetics of the reaction intermediates and transition states for both surface species as active sites. This enables the construction of free-energy diagrams for the Volmer-Heyrovsky and Volmer-Tafel mechanisms and allows for the simulation of Tafel plots by a rigorous microkinetic framework. While the active-site motif V<sub>2</sub>C-OH seems to be less relevant for the HER under typical reaction conditions, we demonstrate that the HER is kinetically facile on the V<sub>2</sub>C-H surface. For this surface termination, we report a potential-depending switching of the preferred mechanism from the Volmer-Heyrovsky to the Volmer-Tafel description with increasing overpotential while encountering similarities to the HER over Pt.</p></div>","PeriodicalId":346,"journal":{"name":"Journal of Catalysis","volume":"421 ","pages":"Pages 252-263"},"PeriodicalIF":6.5000,"publicationDate":"2023-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"4","resultStr":"{\"title\":\"Theoretical study of the mechanism of the hydrogen evolution reaction on the V2C MXene: Thermodynamic and kinetic aspects\",\"authors\":\"Martí López , Kai S. Exner , Francesc Viñes , Francesc Illas\",\"doi\":\"10.1016/j.jcat.2023.03.027\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Both experimentally and theoretically, the MXene family has shown promising hydrogen evolution reaction (HER) capabilities. However, so far, the theoretical approach has been relying on the well-known thermodynamic descriptor, Δ<em>G</em><sub>H</sub>, whereas experimental studies report Tafel plots, containing kinetic rather than thermodynamic information. Aiming to link theory to experiments, the present study explores five different HER pathways over the exemplary V<sub>2</sub>C (0001) MXene by density functional theory calculations. While the surface coverage under HER conditions (with either H* or OH* adsorbates) is extracted from a Pourbaix diagram, we determine the energetics of the reaction intermediates and transition states for both surface species as active sites. This enables the construction of free-energy diagrams for the Volmer-Heyrovsky and Volmer-Tafel mechanisms and allows for the simulation of Tafel plots by a rigorous microkinetic framework. While the active-site motif V<sub>2</sub>C-OH seems to be less relevant for the HER under typical reaction conditions, we demonstrate that the HER is kinetically facile on the V<sub>2</sub>C-H surface. For this surface termination, we report a potential-depending switching of the preferred mechanism from the Volmer-Heyrovsky to the Volmer-Tafel description with increasing overpotential while encountering similarities to the HER over Pt.</p></div>\",\"PeriodicalId\":346,\"journal\":{\"name\":\"Journal of Catalysis\",\"volume\":\"421 \",\"pages\":\"Pages 252-263\"},\"PeriodicalIF\":6.5000,\"publicationDate\":\"2023-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"4\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Catalysis\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0021951723001148\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Catalysis","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0021951723001148","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Theoretical study of the mechanism of the hydrogen evolution reaction on the V2C MXene: Thermodynamic and kinetic aspects
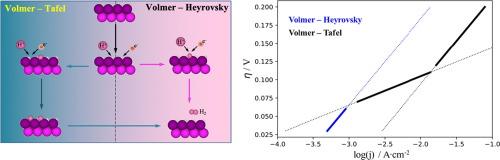
Both experimentally and theoretically, the MXene family has shown promising hydrogen evolution reaction (HER) capabilities. However, so far, the theoretical approach has been relying on the well-known thermodynamic descriptor, ΔGH, whereas experimental studies report Tafel plots, containing kinetic rather than thermodynamic information. Aiming to link theory to experiments, the present study explores five different HER pathways over the exemplary V2C (0001) MXene by density functional theory calculations. While the surface coverage under HER conditions (with either H* or OH* adsorbates) is extracted from a Pourbaix diagram, we determine the energetics of the reaction intermediates and transition states for both surface species as active sites. This enables the construction of free-energy diagrams for the Volmer-Heyrovsky and Volmer-Tafel mechanisms and allows for the simulation of Tafel plots by a rigorous microkinetic framework. While the active-site motif V2C-OH seems to be less relevant for the HER under typical reaction conditions, we demonstrate that the HER is kinetically facile on the V2C-H surface. For this surface termination, we report a potential-depending switching of the preferred mechanism from the Volmer-Heyrovsky to the Volmer-Tafel description with increasing overpotential while encountering similarities to the HER over Pt.
期刊介绍:
The Journal of Catalysis publishes scholarly articles on both heterogeneous and homogeneous catalysis, covering a wide range of chemical transformations. These include various types of catalysis, such as those mediated by photons, plasmons, and electrons. The focus of the studies is to understand the relationship between catalytic function and the underlying chemical properties of surfaces and metal complexes.
The articles in the journal offer innovative concepts and explore the synthesis and kinetics of inorganic solids and homogeneous complexes. Furthermore, they discuss spectroscopic techniques for characterizing catalysts, investigate the interaction of probes and reacting species with catalysts, and employ theoretical methods.
The research presented in the journal should have direct relevance to the field of catalytic processes, addressing either fundamental aspects or applications of catalysis.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


