Kevin Basemann*, Jennifer J. Becker, Theresa L. Windus and Michel R. Gagné*,
{"title":"极性亲电试剂还原消除步骤中的电子重排导致氧化还原事件的细化","authors":"Kevin Basemann*, Jennifer J. Becker, Theresa L. Windus and Michel R. Gagné*, ","doi":"10.1021/acs.organomet.3c00102","DOIUrl":null,"url":null,"abstract":"<p >The oxidative addition/reductive elimination of polar molecules such as methyl iodide at late metal centers has a strongly supported S<sub>N</sub>2 mechanism for many key organometallic complexes, including important industrial catalysts. In the reductive elimination direction, it is proposed that a ligand initially dissociates, typically a halide, followed by subsequent nucleophilic attack at the ligand trans to the now vacant site. The prevailing view is the metal reduction occurs upon transferring the electrophile in the S<sub>N</sub>2 step. Herein, we report the use of an ensemble of computational techniques to characterize the electronic structure of the reactants and intermediates along this reductive elimination pathway. These calculations demonstrate, unexpectedly, that the initiating loss of an anionic ligand from the octahedral highly oxidized structure leads to an electronic rearrangement that shifts electron density from the apical ligand back toward the metal resulting in an inversion of the electron flow between the metal and apical ligand. The anisotropic shift in electron density to the metal disproportionately affects the apical position, which is best described as a Pt → Me dative bond. With this Pt → Me bonding description, our interpretation of the IUPAC oxidation state formalism would assign the intermediate as Pt<sup>II</sup>. Although counterintuitive, the formal and functional reduction of the metal thus occurs upon halide dissociation.</p>","PeriodicalId":56,"journal":{"name":"Organometallics","volume":"42 16","pages":"2171–2176"},"PeriodicalIF":2.9000,"publicationDate":"2023-08-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Electronic Rearrangement in Steps of Reductive Elimination of Polar Electrophiles Leads to Refinement of Redox Events\",\"authors\":\"Kevin Basemann*, Jennifer J. Becker, Theresa L. Windus and Michel R. Gagné*, \",\"doi\":\"10.1021/acs.organomet.3c00102\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The oxidative addition/reductive elimination of polar molecules such as methyl iodide at late metal centers has a strongly supported S<sub>N</sub>2 mechanism for many key organometallic complexes, including important industrial catalysts. In the reductive elimination direction, it is proposed that a ligand initially dissociates, typically a halide, followed by subsequent nucleophilic attack at the ligand trans to the now vacant site. The prevailing view is the metal reduction occurs upon transferring the electrophile in the S<sub>N</sub>2 step. Herein, we report the use of an ensemble of computational techniques to characterize the electronic structure of the reactants and intermediates along this reductive elimination pathway. These calculations demonstrate, unexpectedly, that the initiating loss of an anionic ligand from the octahedral highly oxidized structure leads to an electronic rearrangement that shifts electron density from the apical ligand back toward the metal resulting in an inversion of the electron flow between the metal and apical ligand. The anisotropic shift in electron density to the metal disproportionately affects the apical position, which is best described as a Pt → Me dative bond. With this Pt → Me bonding description, our interpretation of the IUPAC oxidation state formalism would assign the intermediate as Pt<sup>II</sup>. Although counterintuitive, the formal and functional reduction of the metal thus occurs upon halide dissociation.</p>\",\"PeriodicalId\":56,\"journal\":{\"name\":\"Organometallics\",\"volume\":\"42 16\",\"pages\":\"2171–2176\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2023-08-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Organometallics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.organomet.3c00102\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, INORGANIC & NUCLEAR\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Organometallics","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.organomet.3c00102","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
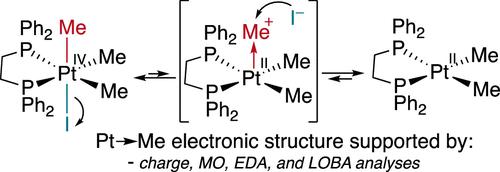
Electronic Rearrangement in Steps of Reductive Elimination of Polar Electrophiles Leads to Refinement of Redox Events
The oxidative addition/reductive elimination of polar molecules such as methyl iodide at late metal centers has a strongly supported SN2 mechanism for many key organometallic complexes, including important industrial catalysts. In the reductive elimination direction, it is proposed that a ligand initially dissociates, typically a halide, followed by subsequent nucleophilic attack at the ligand trans to the now vacant site. The prevailing view is the metal reduction occurs upon transferring the electrophile in the SN2 step. Herein, we report the use of an ensemble of computational techniques to characterize the electronic structure of the reactants and intermediates along this reductive elimination pathway. These calculations demonstrate, unexpectedly, that the initiating loss of an anionic ligand from the octahedral highly oxidized structure leads to an electronic rearrangement that shifts electron density from the apical ligand back toward the metal resulting in an inversion of the electron flow between the metal and apical ligand. The anisotropic shift in electron density to the metal disproportionately affects the apical position, which is best described as a Pt → Me dative bond. With this Pt → Me bonding description, our interpretation of the IUPAC oxidation state formalism would assign the intermediate as PtII. Although counterintuitive, the formal and functional reduction of the metal thus occurs upon halide dissociation.
期刊介绍:
Organometallics is the flagship journal of organometallic chemistry and records progress in one of the most active fields of science, bridging organic and inorganic chemistry. The journal publishes Articles, Communications, Reviews, and Tutorials (instructional overviews) that depict research on the synthesis, structure, bonding, chemical reactivity, and reaction mechanisms for a variety of applications, including catalyst design and catalytic processes; main-group, transition-metal, and lanthanide and actinide metal chemistry; synthetic aspects of polymer science and materials science; and bioorganometallic chemistry.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


