具有主动训练和通用机器学习力场的路径积分分子动力学
IF 3.4
2区 物理与天体物理
Q1 COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS
引用次数: 0
摘要
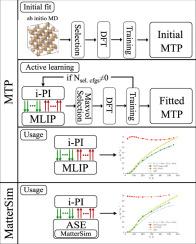
计算核量子效应(NQEs)可以显著改变有限温度下的材料性质。使用路径积分分子动力学(PIMD)方法的原子建模可以完全解释这种效应,但需要计算效率高和原子间相互作用的精确模型。经验势是快速的,但可能缺乏足够的精度,而量子力学计算是高度精确的,但计算昂贵。机器学习原子间势为这一挑战提供了解决方案,与密度泛函理论(DFT)计算相比,它提供了接近量子力学的精度,同时保持了较高的计算效率。在这种情况下,开发了一个接口,使用i-PI软件包(MTP-PIMD方法)将MLIP-2软件包中的矩张量势(MTPs)集成到PIMD计算中。然后将该界面应用于电位的主动学习,并研究NQEs对材料性质的影响,即氢化锂(LiH)和硅(Si)体系中晶格参数和线性晶格热膨胀(LTE)系数的温度依赖性以及径向分布函数。用MTPs得到的LiH和Si的线性LTE对温度的依赖关系与可用的实验依赖关系以及用MatterSim通用机器学习力场和准谐波近似计算的依赖关系很好地一致。因此,MTP-PIMD方法被证明是高度准确和有效的。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Path-integral molecular dynamics with actively-trained and universal machine learning force fields
Accounting for nuclear quantum effects (NQEs) can significantly alter material properties at finite temperatures. Atomic modeling using the path-integral molecular dynamics (PIMD) method can fully account for such effects, but requires computationally efficient and accurate models of interatomic interactions. Empirical potentials are fast but may lack sufficient accuracy, whereas quantum-mechanical calculations are highly accurate but computationally expensive. Machine-learned interatomic potentials offer a solution to this challenge, providing near-quantum-mechanical accuracy while maintaining high computational efficiency compared to density functional theory (DFT) calculations. In this context, an interface was developed to integrate moment tensor potentials (MTPs) from the MLIP-2 software package into PIMD calculations using the i-PI software package (the MTP-PIMD approach). This interface was then applied to active learning of potentials and to investigate the influence of NQEs on material properties, namely the temperature dependence of lattice parameters and linear lattice thermal expansion (LTE) coefficients, as well as radial distribution functions, for lithium hydride (LiH) and silicon (Si) systems. The dependencies of the linear LTE on temperature for LiH and Si obtained with MTPs are in a good agreement with available experimental dependencies and with the ones calculated with the MatterSim universal machine learning force field and with the quasi-harmonic approximation. The MTP-PIMD approach thus proves to be highly accurate and effective.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Computer Physics Communications
物理-计算机:跨学科应用
CiteScore
12.10
自引率
3.20%
发文量
287
审稿时长
5.3 months
期刊介绍:
The focus of CPC is on contemporary computational methods and techniques and their implementation, the effectiveness of which will normally be evidenced by the author(s) within the context of a substantive problem in physics. Within this setting CPC publishes two types of paper.
Computer Programs in Physics (CPiP)
These papers describe significant computer programs to be archived in the CPC Program Library which is held in the Mendeley Data repository. The submitted software must be covered by an approved open source licence. Papers and associated computer programs that address a problem of contemporary interest in physics that cannot be solved by current software are particularly encouraged.
Computational Physics Papers (CP)
These are research papers in, but are not limited to, the following themes across computational physics and related disciplines.
mathematical and numerical methods and algorithms;
computational models including those associated with the design, control and analysis of experiments; and
algebraic computation.
Each will normally include software implementation and performance details. The software implementation should, ideally, be available via GitHub, Zenodo or an institutional repository.In addition, research papers on the impact of advanced computer architecture and special purpose computers on computing in the physical sciences and software topics related to, and of importance in, the physical sciences may be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


