原子无序材料集成特性建模的机器学习框架
IF 16
1区 材料科学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
摘要
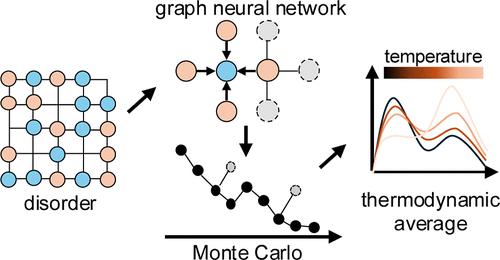
原子无序可以强烈地影响材料的性质,如电荷输运、光学响应和催化活性。然而,由于采样大构型空间和计算复杂物理量的成本,对第一原理方法来说,有效地模拟这些无序效应仍然是一个挑战。机器学习技术的最新进展,特别是图神经网络(gnn),已经能够有效和准确地预测复杂的材料特性,为研究无序系统提供了有前途的工具。在这项工作中,我们提出了一个通用的机器学习辅助计算框架,该框架将等变GNNs与蒙特卡罗模拟集成在一起,以计算无序材料的热力学和集合平均功能特性。以表面末端无序的MXene单层Ti3C2T2-x为代表体系,我们发现由于电子散射和掺杂的相互作用,在有序-无序相变温度附近电导率出现了一个突现峰。相比之下,光学电导率对局部原子无序程度不敏感,反映的是整体表面化学成分。这些结果突出了原子无序在影响材料性能方面的作用,并展示了我们的方法在高熵合金和自旋液体等广泛材料中统计建模无序效应的潜力。本文章由计算机程序翻译,如有差异,请以英文原文为准。
A Machine Learning Framework for Modeling Ensemble Properties of Atomically Disordered Materials
Atomic disorder can strongly influence material properties such as charge transport, optical response, and catalytic activity. However, efficiently modeling these disorder effects remains challenging for first-principles methods due to the cost of sampling large configurational spaces and computing complex physical quantities. Recent advances of machine learning techniques, particularly graph neural networks (GNNs), has enabled the efficient and accurate predictions of complex material properties, offering promising tools for studying disordered systems. In this work, we present a general machine-learning-assisted computational framework that integrates equivariant GNNs with Monte Carlo simulations to compute the thermodynamic and ensemble-averaged functional properties of disordered materials. Using the surface-termination-disordered MXene monolayer Ti3C2T2–x as a representative system, we find that electrical conductivity exhibits an emergent peak near the order–disorder phase transition temperature due to the interplay between electron scattering and doping. In contrast, optical conductivity remains largely insensitive to local atomic disorder and reflects the global surface chemical composition. These results highlight the role of atomic disorder in affecting material properties and demonstrate the potential of our approach for statistically modeling disorder effects in a wide range of materials such as high-entropy alloys and spin liquids.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

ACS Nano
工程技术-材料科学:综合
CiteScore
26.00
自引率
4.10%
发文量
1627
审稿时长
1.7 months
期刊介绍:
ACS Nano, published monthly, serves as an international forum for comprehensive articles on nanoscience and nanotechnology research at the intersections of chemistry, biology, materials science, physics, and engineering. The journal fosters communication among scientists in these communities, facilitating collaboration, new research opportunities, and advancements through discoveries. ACS Nano covers synthesis, assembly, characterization, theory, and simulation of nanostructures, nanobiotechnology, nanofabrication, methods and tools for nanoscience and nanotechnology, and self- and directed-assembly. Alongside original research articles, it offers thorough reviews, perspectives on cutting-edge research, and discussions envisioning the future of nanoscience and nanotechnology.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


