石墨烯作为锂离子电池应用基准材料的DFT和KMC研究
IF 1.8
4区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
近年来,许多研究探索了改性或功能化石墨烯,如掺杂石墨烯或石墨烯复合材料,以提高锂离子电池的性能。相比之下,我们的研究重点是原始石墨烯,结合密度泛函理论(DFT)和动力学蒙特卡罗(KMC)模拟,建立对其内在特性的基本理解。这种方法为未来的材料设计提供了关键的基准。DFT表明,锂优先吸附在具有强结合能(- 1.9 eV)的空心位点,诱导半金属到金属的转变。该材料具有较高的理论容量(744 mAh/g)、中等的平均电压(0.78 V)和低Li扩散势垒(0.31 eV)。KMC模拟进一步量化了浓度和温度对锂扩散率的依赖,得出了锂浓度在[0.01-0.1]范围内的经验关系DLi(CLi,T)。在室温下,计算得到的DLi值范围为2 × 10−9 ~ 5 × 10−8 cm²/s,与实验数据吻合良好。本文章由计算机程序翻译,如有差异,请以英文原文为准。
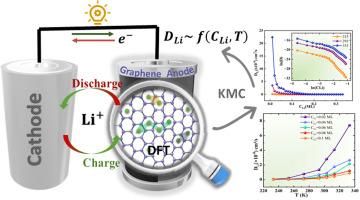
DFT and KMC study of graphene as a benchmark material for Li-Ion battery applications
In recent years, many studies have explored modified or functionalized graphene, such as doped graphene or graphene composites, to enhance Li-ion battery performance. In contrast, our study focuses on pristine graphene to establish a fundamental understanding of its intrinsic properties, combining density functional theory (DFT) and kinetic Monte Carlo (KMC) simulations. This approach provides critical benchmarks for future material design. DFT reveals that lithium preferentially adsorbs at hollow site with a strong binding energy (−1.9 eV), inducing a semi-metal-to-metal transition. The material exhibits a high theoretical capacity (744 mAh/g), a moderate average voltage (0.78 V), and a low Li diffusion barrier (0.31 eV). KMC simulations further quantify the concentration and temperature dependent Li diffusivity, yielding the empirical relation (,T) for Li concentrations in the range [0.01–0.1]. At room temperature, the calculated values span 2 × 10−9 to 5 × 10−8 cm²/s, showing excellent agreement with experimental data.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Surface Science
化学-物理:凝聚态物理
CiteScore
3.30
自引率
5.30%
发文量
137
审稿时长
25 days
期刊介绍:
Surface Science is devoted to elucidating the fundamental aspects of chemistry and physics occurring at a wide range of surfaces and interfaces and to disseminating this knowledge fast. The journal welcomes a broad spectrum of topics, including but not limited to:
• model systems (e.g. in Ultra High Vacuum) under well-controlled reactive conditions
• nanoscale science and engineering, including manipulation of matter at the atomic/molecular scale and assembly phenomena
• reactivity of surfaces as related to various applied areas including heterogeneous catalysis, chemistry at electrified interfaces, and semiconductors functionalization
• phenomena at interfaces relevant to energy storage and conversion, and fuels production and utilization
• surface reactivity for environmental protection and pollution remediation
• interactions at surfaces of soft matter, including polymers and biomaterials.
Both experimental and theoretical work, including modeling, is within the scope of the journal. Work published in Surface Science reaches a wide readership, from chemistry and physics to biology and materials science and engineering, providing an excellent forum for cross-fertilization of ideas and broad dissemination of scientific discoveries.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


