药物发现中的自由能计算
IF 17.7
1区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
摘要
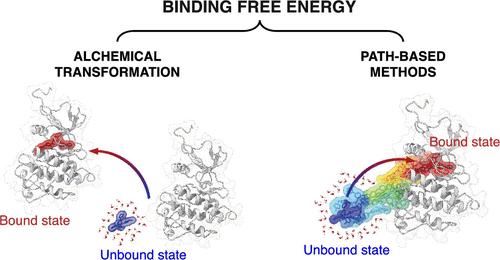
本文讨论了约束自由能计算的最新进展和挑战,重点介绍了两类增强采样技术:炼金术变换和基于路径的方法。结合自由能是药物发现的一个关键指标,因为它测量了配体对其靶受体的亲和力。自由能和亲和力指导潜在候选药物的排序和选择。自由能计算的理论基础在几十年前就建立起来了,但将其有效地应用于药物靶标结合仍然是计算药物设计中的一个巨大挑战。主要的障碍来自采样问题(因为绑定是一个罕见的事件)、力场精度限制和模拟收敛。炼金术变换现在是制药工业中最常用的计算结合自由能的方法。然而,虽然这些方法可以有效地计算能量差,但它们的应用往往局限于相对束缚自由能的计算。绝对准确(误差1千卡/摩尔)的结合自由能预测仍然是计算化学家和物理学家面临的巨大挑战之一。炼金术方法的另一个限制是,它们缺乏提供结合过程的机制或动力学见解的能力,这对于优化先导化合物和设计新疗法至关重要。原则上,基于路径的方法提供了准确估计绝对结合自由能的可能性,同时也提供了对结合途径和相互作用的见解。本文章由计算机程序翻译,如有差异,请以英文原文为准。
On Free Energy Calculations in Drug Discovery
This Account discusses recent progress and challenges in binding free energy computations, focusing on two classes of enhanced sampling techniques: alchemical transformations and path-based methods. Binding free energy is a crucial metric in drug discovery, as it measures the affinity of a ligand for its target receptor. Free energy and affinity guide the ranking and selection of potential drug candidates. The theoretical foundations of free energy calculations were established several decades ago, but their efficient application to drug-target binding remains a grand challenge in computational drug design. The main obstacles stem from sampling issues (as binding is a rare event), force field accuracy limitations, and simulation convergence. Alchemical transformations are now the most used methods for computing binding free energies in the pharmaceutical industry. However, while they efficiently calculate energy differences, the application of these methods is often limited to relative binding free energy calculations. Absolute and accurate (error < 1 kcal/mol) binding free energy predictions remain one of the great challenges for computational chemists and physicists. Another limitation of alchemical methods is that they lack the ability to provide mechanistic or kinetic insights into the binding process, crucial for optimizing lead compounds and designing novel therapies. Path-based methods offer, in principle, the possibility to accurately estimate absolute binding free energy while also providing insights into binding pathways and interactions.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Accounts of Chemical Research
化学-化学综合
CiteScore
31.40
自引率
1.10%
发文量
312
审稿时长
2 months
期刊介绍:
Accounts of Chemical Research presents short, concise and critical articles offering easy-to-read overviews of basic research and applications in all areas of chemistry and biochemistry. These short reviews focus on research from the author’s own laboratory and are designed to teach the reader about a research project. In addition, Accounts of Chemical Research publishes commentaries that give an informed opinion on a current research problem. Special Issues online are devoted to a single topic of unusual activity and significance.
Accounts of Chemical Research replaces the traditional article abstract with an article "Conspectus." These entries synopsize the research affording the reader a closer look at the content and significance of an article. Through this provision of a more detailed description of the article contents, the Conspectus enhances the article's discoverability by search engines and the exposure for the research.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


