Yixin Zhu, Chenxi Lv, Yanheng Qiao, Hanqi Yang, Wentong Lin, Xuchen Wang, Yueqi Zhang, Bo Yang
{"title":"急性肾损伤向慢性肾病的转变:线粒体自噬与NLRP3炎性体的相互作用。","authors":"Yixin Zhu, Chenxi Lv, Yanheng Qiao, Hanqi Yang, Wentong Lin, Xuchen Wang, Yueqi Zhang, Bo Yang","doi":"10.3389/fmolb.2025.1643829","DOIUrl":null,"url":null,"abstract":"<p><p>Acute kidney injury (AKI) and chronic kidney disease (CKD) are closely interrelated renal disorders, where AKI frequently progresses to CKD, resulting in irreversible loss of renal function. In recent years, the roles of the NLRP3 inflammasome and mitophagy in the AKI-to-CKD transition have attracted significant attention. As a crucial component of the innate immune system, the NLRP3 inflammasome promotes AKI-to-CKD progression by mediating inflammatory responses and cellular pyroptosis during renal injury. Conversely, mitophagy exerts renoprotective effects through the selective removal of damaged mitochondria, maintenance of cellular homeostasis, and alleviation of inflammation and oxidative stress. Studies demonstrate that NLRP3 activation is closely associated with mitochondrial dysfunction, while mitophagy can suppress NLRP3 activation by clearing damaged mitochondria, establishing a negative feedback regulatory mechanism. During the AKI phase, mitochondrial damage and excessive NLRP3 activation exacerbate renal tubular epithelial cell injury and inflammatory responses. Concurrently, persistent NLRP3 activation and impaired mitophagy lead to chronic inflammation and fibrosis, accelerating the transition from AKI to CKD. Therefore, targeting the NLRP3 inflammasome and modulating mitophagy may emerge as novel therapeutic strategies for AKI-to-CKD transition. This review focuses on elucidating the molecular mechanisms between mitophagy and the NLRP3 inflammasome, along with related targeted therapies, to provide new insights for preventing AKI progression to CKD.</p>","PeriodicalId":12465,"journal":{"name":"Frontiers in Molecular Biosciences","volume":"12 ","pages":"1643829"},"PeriodicalIF":3.9000,"publicationDate":"2025-09-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12504097/pdf/","citationCount":"0","resultStr":"{\"title\":\"Transformation of acute kidney injury to chronic kidney disease: the interaction between mitophagy and NLRP3 inflammasome.\",\"authors\":\"Yixin Zhu, Chenxi Lv, Yanheng Qiao, Hanqi Yang, Wentong Lin, Xuchen Wang, Yueqi Zhang, Bo Yang\",\"doi\":\"10.3389/fmolb.2025.1643829\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Acute kidney injury (AKI) and chronic kidney disease (CKD) are closely interrelated renal disorders, where AKI frequently progresses to CKD, resulting in irreversible loss of renal function. In recent years, the roles of the NLRP3 inflammasome and mitophagy in the AKI-to-CKD transition have attracted significant attention. As a crucial component of the innate immune system, the NLRP3 inflammasome promotes AKI-to-CKD progression by mediating inflammatory responses and cellular pyroptosis during renal injury. Conversely, mitophagy exerts renoprotective effects through the selective removal of damaged mitochondria, maintenance of cellular homeostasis, and alleviation of inflammation and oxidative stress. Studies demonstrate that NLRP3 activation is closely associated with mitochondrial dysfunction, while mitophagy can suppress NLRP3 activation by clearing damaged mitochondria, establishing a negative feedback regulatory mechanism. During the AKI phase, mitochondrial damage and excessive NLRP3 activation exacerbate renal tubular epithelial cell injury and inflammatory responses. Concurrently, persistent NLRP3 activation and impaired mitophagy lead to chronic inflammation and fibrosis, accelerating the transition from AKI to CKD. Therefore, targeting the NLRP3 inflammasome and modulating mitophagy may emerge as novel therapeutic strategies for AKI-to-CKD transition. This review focuses on elucidating the molecular mechanisms between mitophagy and the NLRP3 inflammasome, along with related targeted therapies, to provide new insights for preventing AKI progression to CKD.</p>\",\"PeriodicalId\":12465,\"journal\":{\"name\":\"Frontiers in Molecular Biosciences\",\"volume\":\"12 \",\"pages\":\"1643829\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2025-09-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12504097/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Frontiers in Molecular Biosciences\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.3389/fmolb.2025.1643829\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in Molecular Biosciences","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.3389/fmolb.2025.1643829","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Transformation of acute kidney injury to chronic kidney disease: the interaction between mitophagy and NLRP3 inflammasome.
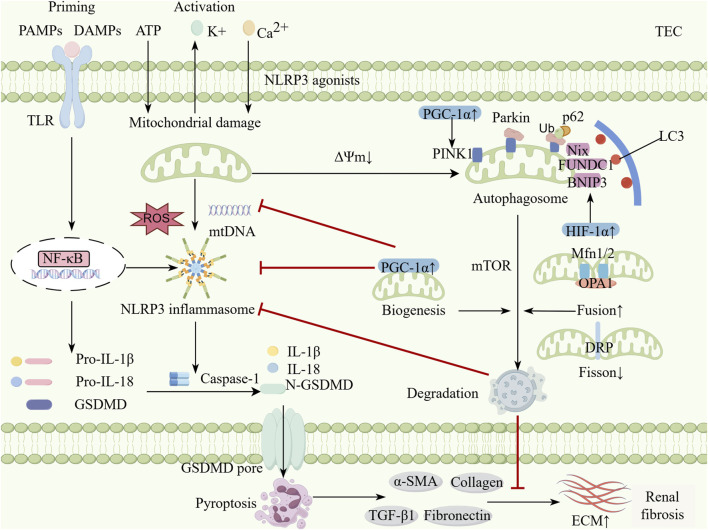
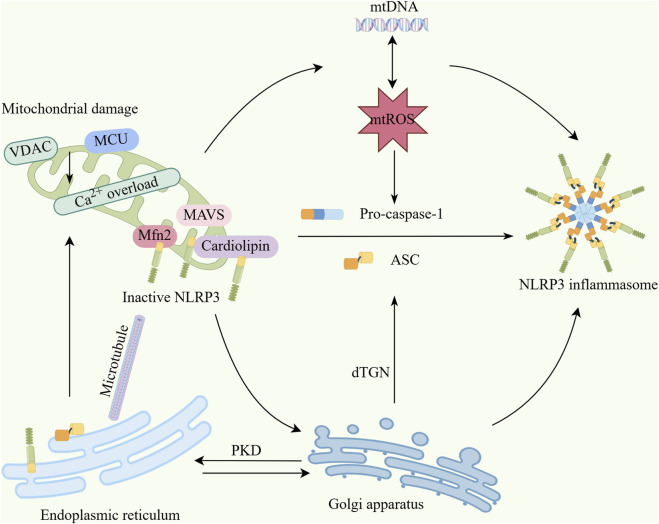
Acute kidney injury (AKI) and chronic kidney disease (CKD) are closely interrelated renal disorders, where AKI frequently progresses to CKD, resulting in irreversible loss of renal function. In recent years, the roles of the NLRP3 inflammasome and mitophagy in the AKI-to-CKD transition have attracted significant attention. As a crucial component of the innate immune system, the NLRP3 inflammasome promotes AKI-to-CKD progression by mediating inflammatory responses and cellular pyroptosis during renal injury. Conversely, mitophagy exerts renoprotective effects through the selective removal of damaged mitochondria, maintenance of cellular homeostasis, and alleviation of inflammation and oxidative stress. Studies demonstrate that NLRP3 activation is closely associated with mitochondrial dysfunction, while mitophagy can suppress NLRP3 activation by clearing damaged mitochondria, establishing a negative feedback regulatory mechanism. During the AKI phase, mitochondrial damage and excessive NLRP3 activation exacerbate renal tubular epithelial cell injury and inflammatory responses. Concurrently, persistent NLRP3 activation and impaired mitophagy lead to chronic inflammation and fibrosis, accelerating the transition from AKI to CKD. Therefore, targeting the NLRP3 inflammasome and modulating mitophagy may emerge as novel therapeutic strategies for AKI-to-CKD transition. This review focuses on elucidating the molecular mechanisms between mitophagy and the NLRP3 inflammasome, along with related targeted therapies, to provide new insights for preventing AKI progression to CKD.
期刊介绍:
Much of contemporary investigation in the life sciences is devoted to the molecular-scale understanding of the relationships between genes and the environment — in particular, dynamic alterations in the levels, modifications, and interactions of cellular effectors, including proteins. Frontiers in Molecular Biosciences offers an international publication platform for basic as well as applied research; we encourage contributions spanning both established and emerging areas of biology. To this end, the journal draws from empirical disciplines such as structural biology, enzymology, biochemistry, and biophysics, capitalizing as well on the technological advancements that have enabled metabolomics and proteomics measurements in massively parallel throughput, and the development of robust and innovative computational biology strategies. We also recognize influences from medicine and technology, welcoming studies in molecular genetics, molecular diagnostics and therapeutics, and nanotechnology.
Our ultimate objective is the comprehensive illustration of the molecular mechanisms regulating proteins, nucleic acids, carbohydrates, lipids, and small metabolites in organisms across all branches of life.
In addition to interesting new findings, techniques, and applications, Frontiers in Molecular Biosciences will consider new testable hypotheses to inspire different perspectives and stimulate scientific dialogue. The integration of in silico, in vitro, and in vivo approaches will benefit endeavors across all domains of the life sciences.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


