反渗透自由基在硝酸酯自催化分解中的关键作用
IF 13.2
1区 工程技术
Q1 ENGINEERING, CHEMICAL
引用次数: 0
摘要
硝酸酯(R-ONO2)由于其优异的能量性能,长期以来一直是固体推进剂的重要成分。然而,它们固有的自催化分解倾向破坏了储存和运行的安全性。虽然NO2自由基-由初始RO-NO2键裂解形成-被认为驱动低温自催化,但其确切机制仍有争议。在这里,我们将分子动力学模拟与量子化学计算相结合,以阐明早期分解事件和维持自催化的关键双分子反应。我们分别选择硝酸异丙酯(iPN)和硝酸甘油(NG)作为模型单硝酸盐和聚硝酸盐酯。结果表明,在iPN的冷凝相热分解中,RO自由基表现出最低的自由能垒和最大的放热性,而不是no2介导的h萃取或酸水解途径。对于NG, RO自由基优先进行单分子解离或与NO2反应,从而传播自由基链途径。这些发现完善了我们对硝酸酯自催化的机理理解,并突出了迄今为止未被充分认识的反渗透自由基的作用。这些见解可能会为硝酸酯基含能材料更有效的稳定剂的合理设计提供信息。本文章由计算机程序翻译,如有差异,请以英文原文为准。
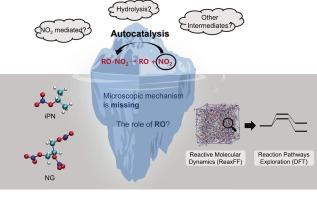
Critical role of RO radicals in autocatalytic decomposition of nitrate esters
Nitrate esters (R–ONO2) have long served as essential constituents in solid propellants owing to their excellent energetic performance. However, their intrinsic tendency toward autocatalytic decomposition undermines storage and operational safety. Although NO2 radicals—formed by initial RO–NO2 bond cleavage—are acknowledged to drive low-temperature autocatalysis, the exact mechanism is disputed. Here, we integrate molecular dynamics simulations with quantum chemical calculations to elucidate the early decomposition events and key bimolecular reactions that sustain autocatalysis. We select isopropyl nitrate (iPN) and nitroglycerin (NG) as model mono- and polynitrate esters, respectively. The results indicate that, in condensed-phase thermal decomposition of iPN, RO radicals—rather than NO2-mediated H-abstraction or acid-hydrolysis pathways—exhibit the lowest free energy barriers and greatest exothermicity. For NG, RO radicals preferentially undergo unimolecular dissociation or react with NO2, thereby propagating radical chain pathways. These findings refine our mechanistic understanding of nitrate ester autocatalysis and highlight the hitherto underappreciated role of RO radicals. Such insights may inform the rational design of more effective stabilizers for nitrate ester-based energetic materials.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Chemical Engineering Journal
工程技术-工程:化工
CiteScore
21.70
自引率
9.30%
发文量
6781
审稿时长
2.4 months
期刊介绍:
The Chemical Engineering Journal is an international research journal that invites contributions of original and novel fundamental research. It aims to provide an international platform for presenting original fundamental research, interpretative reviews, and discussions on new developments in chemical engineering. The journal welcomes papers that describe novel theory and its practical application, as well as those that demonstrate the transfer of techniques from other disciplines. It also welcomes reports on carefully conducted experimental work that is soundly interpreted. The main focus of the journal is on original and rigorous research results that have broad significance. The Catalysis section within the Chemical Engineering Journal focuses specifically on Experimental and Theoretical studies in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. These studies have industrial impact on various sectors such as chemicals, energy, materials, foods, healthcare, and environmental protection.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


