SARS-CoV-2 n7 -甲基转移酶抑制剂:选择性和强效抗病毒药物
IF 4.7
3区 医学
Q1 PHARMACOLOGY & PHARMACY
引用次数: 0
摘要
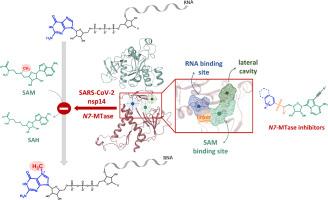
最近的研究已经确定nsp14 n7 -甲基转移酶(N7-MTase)是开发针对SARS-CoV-2的新型抗病毒药物的有希望的治疗靶点。N7-MTase利用s -腺苷- l-蛋氨酸(SAM)作为甲基供体,介导病毒RNA盖帽的第一步甲基化,这是SARS-CoV-2复制和免疫逃逸所必需的。为了设计冠状病毒nsp14 N7-MTase的选择性和有效抑制剂,各个研究小组都将重点放在了SAM的nsp14结合位点上。本文分析了迄今为止设计的有前途的冠状病毒N7-MTase抑制剂,特别关注SAM/ s -腺苷- l-同型半胱氨酸(SAH)类似物,这些类似物可以进一步扩展到占据RNA结合位点和/或邻近的侧腔。研究了抑制剂的构效关系(SAR)数据和结合模式。本研究强调了目前阻碍有效抗病毒药物开发的局限性,特别是有限的选择性和细胞活性,并讨论了解决这些问题的潜在策略。特别是,c -核苷的设计已经显示出有希望的结果,尽管还没有抑制剂进入临床试验。因此,需要进一步努力来确定可行的候选药物。本文章由计算机程序翻译,如有差异,请以英文原文为准。
SARS-CoV-2 N7-methyltransferase inhibitors: Towards selective and potent antivirals
Recent studies have identified nsp14 N7-methyltransferase (N7-MTase) as a promising therapeutic target for the development of new antiviral agents against SARS-CoV-2. Utilising S-adenosyl-L-methionine (SAM) as a methyl donor, N7-MTase mediates the first methylation step in viral RNA capping, which is necessary for the replication of SARS-CoV-2 and its immune evasion. To design selective and potent inhibitors of CoV nsp14 N7-MTase, various research groups have focused on targeting the nsp14 binding site for SAM. In this paper, promising CoV N7-MTase inhibitors designed to date are analysed with a particular focus on SAM/S-adenosyl-L-homocysteine (SAH) analogues, which can be further extended to occupy the RNA binding site and/or the adjacent lateral cavity. The structure-activity relationship (SAR) data and binding modes of the inhibitors are also investigated. This study highlights limitations that currently hinder the development of effective antiviral agents, notably limited selectivity and cellular activity, and discusses potential strategies to address them. In particular, the design of C-nucleosides has shown promising results, although no inhibitor has reached clinical trials yet. Thus, further efforts are necessary to identify viable drug candidates.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
9.60
自引率
2.20%
发文量
248
审稿时长
50 days
期刊介绍:
The journal publishes research articles, review articles and scientific commentaries on all aspects of the pharmaceutical sciences with emphasis on conceptual novelty and scientific quality. The Editors welcome articles in this multidisciplinary field, with a focus on topics relevant for drug discovery and development.
More specifically, the Journal publishes reports on medicinal chemistry, pharmacology, drug absorption and metabolism, pharmacokinetics and pharmacodynamics, pharmaceutical and biomedical analysis, drug delivery (including gene delivery), drug targeting, pharmaceutical technology, pharmaceutical biotechnology and clinical drug evaluation. The journal will typically not give priority to manuscripts focusing primarily on organic synthesis, natural products, adaptation of analytical approaches, or discussions pertaining to drug policy making.
Scientific commentaries and review articles are generally by invitation only or by consent of the Editors. Proceedings of scientific meetings may be published as special issues or supplements to the Journal.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


