{"title":"GPU加速DFT交换-相关泛函求值的高效算法。","authors":"Ryan Stocks, Giuseppe M J Barca","doi":"10.1021/acs.jctc.5c01229","DOIUrl":null,"url":null,"abstract":"<p><p>Kohn-Sham density functional theory (KS-DFT) has become a cornerstone for studying the electronic structure of molecules and materials. Improving algorithmic efficiency through hardware-aware implementations enables application to larger systems and more efficient generation of larger training data sets for machine-learning. In this work, we present a comparative study of four GPU-accelerated algorithms for evaluating the KS-DFT exchange-correlation (XC) potential with an atom-centered Gaussian basis. Two approaches, both leveraging batched dense linear algebra, are found to outperform the others across a suite of molecular benchmarks. We show that batched formation of the XC matrix from the density matrix yields the best performance for large (<math><mo>></mo><mi>O</mi><mrow><mo>(</mo><msup><mn>10</mn><mn>3</mn></msup><mo>)</mo></mrow></math> basis functions), sparse systems such as glycine chains and water clusters. In contrast, for smaller and denser systems such as diamond nanoparticles, especially if employing large basis sets, algorithms that use the underlying molecular orbital coefficients offer superior performance, despite their higher formal scaling. Our implementations deliver speedups of 1.4-5.2× for XC potential evaluation relative to leading GPU-accelerated KS-DFT codes, significantly lowering the computational cost and enabling the routine use of larger integration grids. Finally, we outline directions for continued performance improvements in light of emerging GPU architectures with emphasis on utilizing mixed-precision capabilities.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2025-10-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Efficient Algorithms for GPU Accelerated Evaluation of the DFT Exchange-Correlation Functional.\",\"authors\":\"Ryan Stocks, Giuseppe M J Barca\",\"doi\":\"10.1021/acs.jctc.5c01229\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Kohn-Sham density functional theory (KS-DFT) has become a cornerstone for studying the electronic structure of molecules and materials. Improving algorithmic efficiency through hardware-aware implementations enables application to larger systems and more efficient generation of larger training data sets for machine-learning. In this work, we present a comparative study of four GPU-accelerated algorithms for evaluating the KS-DFT exchange-correlation (XC) potential with an atom-centered Gaussian basis. Two approaches, both leveraging batched dense linear algebra, are found to outperform the others across a suite of molecular benchmarks. We show that batched formation of the XC matrix from the density matrix yields the best performance for large (<math><mo>></mo><mi>O</mi><mrow><mo>(</mo><msup><mn>10</mn><mn>3</mn></msup><mo>)</mo></mrow></math> basis functions), sparse systems such as glycine chains and water clusters. In contrast, for smaller and denser systems such as diamond nanoparticles, especially if employing large basis sets, algorithms that use the underlying molecular orbital coefficients offer superior performance, despite their higher formal scaling. Our implementations deliver speedups of 1.4-5.2× for XC potential evaluation relative to leading GPU-accelerated KS-DFT codes, significantly lowering the computational cost and enabling the routine use of larger integration grids. Finally, we outline directions for continued performance improvements in light of emerging GPU architectures with emphasis on utilizing mixed-precision capabilities.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-10-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.5c01229\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.5c01229","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Efficient Algorithms for GPU Accelerated Evaluation of the DFT Exchange-Correlation Functional.
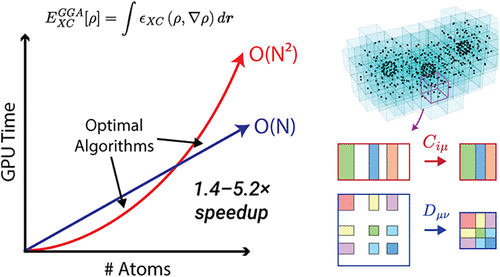
Kohn-Sham density functional theory (KS-DFT) has become a cornerstone for studying the electronic structure of molecules and materials. Improving algorithmic efficiency through hardware-aware implementations enables application to larger systems and more efficient generation of larger training data sets for machine-learning. In this work, we present a comparative study of four GPU-accelerated algorithms for evaluating the KS-DFT exchange-correlation (XC) potential with an atom-centered Gaussian basis. Two approaches, both leveraging batched dense linear algebra, are found to outperform the others across a suite of molecular benchmarks. We show that batched formation of the XC matrix from the density matrix yields the best performance for large ( basis functions), sparse systems such as glycine chains and water clusters. In contrast, for smaller and denser systems such as diamond nanoparticles, especially if employing large basis sets, algorithms that use the underlying molecular orbital coefficients offer superior performance, despite their higher formal scaling. Our implementations deliver speedups of 1.4-5.2× for XC potential evaluation relative to leading GPU-accelerated KS-DFT codes, significantly lowering the computational cost and enabling the routine use of larger integration grids. Finally, we outline directions for continued performance improvements in light of emerging GPU architectures with emphasis on utilizing mixed-precision capabilities.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


