{"title":"量子化学中的混合张量网络和神经网络量子态。","authors":"Zibo Wu, Bohan Zhang, Wei-Hai Fang, Zhendong Li","doi":"10.1021/acs.jctc.5c01228","DOIUrl":null,"url":null,"abstract":"<p><p>Neural network quantum states (NQS) have emerged as a powerful and flexible framework for addressing quantum many-body problems. While successful for model Hamiltonians, their application to molecular systems remains challenging for several reasons. In this work, we introduce three innovations to overcome some of the key limitations. (1) We develop a bounded-degree graph recurrent neural network (BDG-RNN) ansatz, which hybridizes the tensor network and neural network states and is more suitable to molecular electronic structure problems. As matrix product states (MPS) can be embedded into this ansatz, good initialization is possible for complex systems. (2) We introduce neural network correlators (NNCs) to further enhance expressivity and improve accuracy, without dramatically modifying the underlying variational Monte Carlo (VMC) optimization framework. Specifically, we introduce two types of restricted Boltzmann machine (RBM)-inspired correlators, namely, cos-RBM and Ising-RBM, which unlike previous correlators, such as Jastrow and real RBM, can adjust the sign structure of the wave function. (3) We introduce a semistochastic algorithm for local energy evaluation, which significantly reduces computational cost while maintaining high accuracy. Combining these advances, we demonstrate that our approaches can achieve chemical accuracy in challenging systems, including the one-dimensional hydrogen chain H<sub>50</sub>, the iron-sulfur cluster [Fe<sub>2</sub>S<sub>2</sub>(SCH<sub>3</sub>)<sub>4</sub>]<sup>2-</sup>, and a three-dimensional 3 × 3 × 2 hydrogen cluster H<sub>18</sub>. These methods are implemented in an open-source package, PyNQS (https://github.com/Quantum-Chemistry-Group-BNU/PyNQS), to advance NQS methodologies for quantum chemistry.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2025-10-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Hybrid Tensor Network and Neural Network Quantum States for Quantum Chemistry.\",\"authors\":\"Zibo Wu, Bohan Zhang, Wei-Hai Fang, Zhendong Li\",\"doi\":\"10.1021/acs.jctc.5c01228\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Neural network quantum states (NQS) have emerged as a powerful and flexible framework for addressing quantum many-body problems. While successful for model Hamiltonians, their application to molecular systems remains challenging for several reasons. In this work, we introduce three innovations to overcome some of the key limitations. (1) We develop a bounded-degree graph recurrent neural network (BDG-RNN) ansatz, which hybridizes the tensor network and neural network states and is more suitable to molecular electronic structure problems. As matrix product states (MPS) can be embedded into this ansatz, good initialization is possible for complex systems. (2) We introduce neural network correlators (NNCs) to further enhance expressivity and improve accuracy, without dramatically modifying the underlying variational Monte Carlo (VMC) optimization framework. Specifically, we introduce two types of restricted Boltzmann machine (RBM)-inspired correlators, namely, cos-RBM and Ising-RBM, which unlike previous correlators, such as Jastrow and real RBM, can adjust the sign structure of the wave function. (3) We introduce a semistochastic algorithm for local energy evaluation, which significantly reduces computational cost while maintaining high accuracy. Combining these advances, we demonstrate that our approaches can achieve chemical accuracy in challenging systems, including the one-dimensional hydrogen chain H<sub>50</sub>, the iron-sulfur cluster [Fe<sub>2</sub>S<sub>2</sub>(SCH<sub>3</sub>)<sub>4</sub>]<sup>2-</sup>, and a three-dimensional 3 × 3 × 2 hydrogen cluster H<sub>18</sub>. These methods are implemented in an open-source package, PyNQS (https://github.com/Quantum-Chemistry-Group-BNU/PyNQS), to advance NQS methodologies for quantum chemistry.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-10-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.5c01228\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.5c01228","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Hybrid Tensor Network and Neural Network Quantum States for Quantum Chemistry.
Neural network quantum states (NQS) have emerged as a powerful and flexible framework for addressing quantum many-body problems. While successful for model Hamiltonians, their application to molecular systems remains challenging for several reasons. In this work, we introduce three innovations to overcome some of the key limitations. (1) We develop a bounded-degree graph recurrent neural network (BDG-RNN) ansatz, which hybridizes the tensor network and neural network states and is more suitable to molecular electronic structure problems. As matrix product states (MPS) can be embedded into this ansatz, good initialization is possible for complex systems. (2) We introduce neural network correlators (NNCs) to further enhance expressivity and improve accuracy, without dramatically modifying the underlying variational Monte Carlo (VMC) optimization framework. Specifically, we introduce two types of restricted Boltzmann machine (RBM)-inspired correlators, namely, cos-RBM and Ising-RBM, which unlike previous correlators, such as Jastrow and real RBM, can adjust the sign structure of the wave function. (3) We introduce a semistochastic algorithm for local energy evaluation, which significantly reduces computational cost while maintaining high accuracy. Combining these advances, we demonstrate that our approaches can achieve chemical accuracy in challenging systems, including the one-dimensional hydrogen chain H50, the iron-sulfur cluster [Fe2S2(SCH3)4]2-, and a three-dimensional 3 × 3 × 2 hydrogen cluster H18. These methods are implemented in an open-source package, PyNQS (https://github.com/Quantum-Chemistry-Group-BNU/PyNQS), to advance NQS methodologies for quantum chemistry.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.
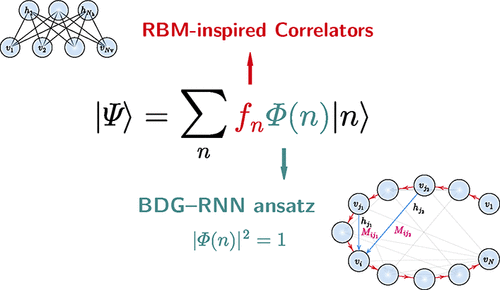

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


