Fatemeh Fathi Niazi, Seungmin Yoon, Khadim Mbacke, Alex Dickson
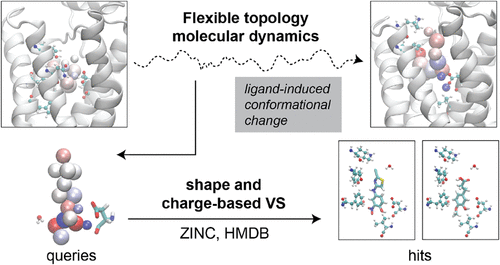
{"title":"柔性拓扑绑定口袋的无向探索。","authors":"Fatemeh Fathi Niazi, Seungmin Yoon, Khadim Mbacke, Alex Dickson","doi":"10.1021/acs.jctc.5c00825","DOIUrl":null,"url":null,"abstract":"<p><p>A common first step in drug design is virtual high-throughput screening (VHTS), where a large number of potential drug molecules are computationally modeled in a protein binding pocket and filtered down to a smaller set of hits that can be further tested computationally or experimentally. Traditional strategies for VHTS do not account for ligand-induced conformational changes in proteins as they typically rely on a single static structure to represent the protein. This neglects the role of binding entropy and the fact that different ligand molecules can induce slightly different conformations in the protein binding site that significantly affect the assessment of a given molecule's fit. To address this challenge, we have developed a method called \"flexible topology\", where a subset of atoms, typically representing a small molecule ligand, can continuously change their atomic identities, which are encoded by a set of attributes that parametrize the nonbonded interactions. These attributes are all implemented as dynamic variables that have masses and evolve over time using gradients of the energy function. In other words, the attributes feel forces from their surrounding environment and respond accordingly. In this way, by observing a set of flexible topology particles move and change in a ligand-binding site, we can learn the preferences of a binding pocket. Here, we demonstrate how undirected flexible topology simulations can be used to explore ligand-binding sites and reveal the desirable properties of potential ligands. We use the β-2-adrenergic receptor as an illustrative example and compare the properties of flexible topology particle groups with a set of 29 B2AR ligand-bound crystal structures, covering 13 distinct ligands. We also show how the shape- and electrostatics-based virtual screening software \"eon\" from OpenEye can be used to find hits that come as close as possible to mimicking the orientation of our flexible topology atoms.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2025-10-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Undirected Exploration of Binding Pockets with Flexible Topology.\",\"authors\":\"Fatemeh Fathi Niazi, Seungmin Yoon, Khadim Mbacke, Alex Dickson\",\"doi\":\"10.1021/acs.jctc.5c00825\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>A common first step in drug design is virtual high-throughput screening (VHTS), where a large number of potential drug molecules are computationally modeled in a protein binding pocket and filtered down to a smaller set of hits that can be further tested computationally or experimentally. Traditional strategies for VHTS do not account for ligand-induced conformational changes in proteins as they typically rely on a single static structure to represent the protein. This neglects the role of binding entropy and the fact that different ligand molecules can induce slightly different conformations in the protein binding site that significantly affect the assessment of a given molecule's fit. To address this challenge, we have developed a method called \\\"flexible topology\\\", where a subset of atoms, typically representing a small molecule ligand, can continuously change their atomic identities, which are encoded by a set of attributes that parametrize the nonbonded interactions. These attributes are all implemented as dynamic variables that have masses and evolve over time using gradients of the energy function. In other words, the attributes feel forces from their surrounding environment and respond accordingly. In this way, by observing a set of flexible topology particles move and change in a ligand-binding site, we can learn the preferences of a binding pocket. Here, we demonstrate how undirected flexible topology simulations can be used to explore ligand-binding sites and reveal the desirable properties of potential ligands. We use the β-2-adrenergic receptor as an illustrative example and compare the properties of flexible topology particle groups with a set of 29 B2AR ligand-bound crystal structures, covering 13 distinct ligands. We also show how the shape- and electrostatics-based virtual screening software \\\"eon\\\" from OpenEye can be used to find hits that come as close as possible to mimicking the orientation of our flexible topology atoms.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-10-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.5c00825\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.5c00825","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Undirected Exploration of Binding Pockets with Flexible Topology.
A common first step in drug design is virtual high-throughput screening (VHTS), where a large number of potential drug molecules are computationally modeled in a protein binding pocket and filtered down to a smaller set of hits that can be further tested computationally or experimentally. Traditional strategies for VHTS do not account for ligand-induced conformational changes in proteins as they typically rely on a single static structure to represent the protein. This neglects the role of binding entropy and the fact that different ligand molecules can induce slightly different conformations in the protein binding site that significantly affect the assessment of a given molecule's fit. To address this challenge, we have developed a method called "flexible topology", where a subset of atoms, typically representing a small molecule ligand, can continuously change their atomic identities, which are encoded by a set of attributes that parametrize the nonbonded interactions. These attributes are all implemented as dynamic variables that have masses and evolve over time using gradients of the energy function. In other words, the attributes feel forces from their surrounding environment and respond accordingly. In this way, by observing a set of flexible topology particles move and change in a ligand-binding site, we can learn the preferences of a binding pocket. Here, we demonstrate how undirected flexible topology simulations can be used to explore ligand-binding sites and reveal the desirable properties of potential ligands. We use the β-2-adrenergic receptor as an illustrative example and compare the properties of flexible topology particle groups with a set of 29 B2AR ligand-bound crystal structures, covering 13 distinct ligands. We also show how the shape- and electrostatics-based virtual screening software "eon" from OpenEye can be used to find hits that come as close as possible to mimicking the orientation of our flexible topology atoms.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


