Wael Shehta, Basant Farag, Shaker Youssif, Samar El-Kalyoubi and Sherin M. Elfeky
{"title":"作为EGFR TK抑制剂和抗增殖剂的氨基嘧啶杂合体的合成、硅和体外研究。","authors":"Wael Shehta, Basant Farag, Shaker Youssif, Samar El-Kalyoubi and Sherin M. Elfeky","doi":"10.1039/D5RA02524A","DOIUrl":null,"url":null,"abstract":"<p >In this article, two series of pyrimidine hybrids, 2-((4-amino-6-(1,3-dioxoisoindolin-2-yl)pyrimidin-2-yl)thio)-<em>N</em>′-(alkylbenzylidene)acetohydrazides (<strong>6a–g</strong>) (Series <strong>I</strong>) and 6-amino-5-((alkylamino)(phenyl)methyl)pyrimidine-2,4(1<em>H</em>,3<em>H</em>)-diones (<strong>10a–d</strong>) (Series <strong>II</strong>), were synthesized using catalyst-free condensation of acid hydrazide <strong>4</strong> with different aromatic aldehydes and nucleophilic substitution reactions of chloromethylpyrimidine with different aliphatic amines , respectively. Compared to gefitinib (IC<small><sub>50</sub></small> = 4.1 ± 0.01 μM) and thalidomide (IC<small><sub>50</sub></small> = 13.4 ± 0.5 μM), compounds <strong>6c</strong> and <strong>10b</strong> exhibited moderate anti-proliferative activity against the MCF-7 breast cancer cell line, with IC<small><sub>50</sub></small> values of 37.7 ± 3.6 and 31.8 ± 2.0 μM, respectively. They demonstrated selective cytotoxicity toward MCF-7 cells over normal WI38 fibroblasts, with IC<small><sub>50</sub></small> values of 87.3 ± 2.6 μM (<strong>6c</strong>) and >100 μM (<strong>10b</strong>). Both compounds showed potent <em>in vitro</em> EGFR-TK enzyme inhibition, with IC<small><sub>50</sub></small> values of 0.9 ± 0.03 μM (<strong>6c</strong>) and 0.7 ± 0.02 μM (<strong>10b</strong>). Molecular docking simulations revealed that <strong>6c</strong> and <strong>10b</strong> effectively bind to the EGFR ATP-binding site, forming hydrogen bonds with the key hinge region amino acid Met793, similar to gefitinib. <em>In silico</em> studies confirmed that both compounds fit the criteria for orally bioavailable drug candidates, showing high gastrointestinal absorption and no predicted adverse effects on the central nervous system or liver. Additionally, both compounds were predicted to be non-carcinogenic and non-mutagenic.</p>","PeriodicalId":102,"journal":{"name":"RSC Advances","volume":" 44","pages":" 36895-36906"},"PeriodicalIF":4.6000,"publicationDate":"2025-10-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12498222/pdf/","citationCount":"0","resultStr":"{\"title\":\"Synthesis, in silico and in vitro studies of aminopyrimidine hybrids as EGFR TK inhibitors and anti-proliferative agents\",\"authors\":\"Wael Shehta, Basant Farag, Shaker Youssif, Samar El-Kalyoubi and Sherin M. Elfeky\",\"doi\":\"10.1039/D5RA02524A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >In this article, two series of pyrimidine hybrids, 2-((4-amino-6-(1,3-dioxoisoindolin-2-yl)pyrimidin-2-yl)thio)-<em>N</em>′-(alkylbenzylidene)acetohydrazides (<strong>6a–g</strong>) (Series <strong>I</strong>) and 6-amino-5-((alkylamino)(phenyl)methyl)pyrimidine-2,4(1<em>H</em>,3<em>H</em>)-diones (<strong>10a–d</strong>) (Series <strong>II</strong>), were synthesized using catalyst-free condensation of acid hydrazide <strong>4</strong> with different aromatic aldehydes and nucleophilic substitution reactions of chloromethylpyrimidine with different aliphatic amines , respectively. Compared to gefitinib (IC<small><sub>50</sub></small> = 4.1 ± 0.01 μM) and thalidomide (IC<small><sub>50</sub></small> = 13.4 ± 0.5 μM), compounds <strong>6c</strong> and <strong>10b</strong> exhibited moderate anti-proliferative activity against the MCF-7 breast cancer cell line, with IC<small><sub>50</sub></small> values of 37.7 ± 3.6 and 31.8 ± 2.0 μM, respectively. They demonstrated selective cytotoxicity toward MCF-7 cells over normal WI38 fibroblasts, with IC<small><sub>50</sub></small> values of 87.3 ± 2.6 μM (<strong>6c</strong>) and >100 μM (<strong>10b</strong>). Both compounds showed potent <em>in vitro</em> EGFR-TK enzyme inhibition, with IC<small><sub>50</sub></small> values of 0.9 ± 0.03 μM (<strong>6c</strong>) and 0.7 ± 0.02 μM (<strong>10b</strong>). Molecular docking simulations revealed that <strong>6c</strong> and <strong>10b</strong> effectively bind to the EGFR ATP-binding site, forming hydrogen bonds with the key hinge region amino acid Met793, similar to gefitinib. <em>In silico</em> studies confirmed that both compounds fit the criteria for orally bioavailable drug candidates, showing high gastrointestinal absorption and no predicted adverse effects on the central nervous system or liver. Additionally, both compounds were predicted to be non-carcinogenic and non-mutagenic.</p>\",\"PeriodicalId\":102,\"journal\":{\"name\":\"RSC Advances\",\"volume\":\" 44\",\"pages\":\" 36895-36906\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2025-10-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12498222/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"RSC Advances\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/ra/d5ra02524a\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"RSC Advances","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/ra/d5ra02524a","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Synthesis, in silico and in vitro studies of aminopyrimidine hybrids as EGFR TK inhibitors and anti-proliferative agents
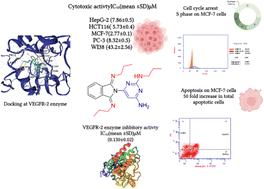
In this article, two series of pyrimidine hybrids, 2-((4-amino-6-(1,3-dioxoisoindolin-2-yl)pyrimidin-2-yl)thio)-N′-(alkylbenzylidene)acetohydrazides (6a–g) (Series I) and 6-amino-5-((alkylamino)(phenyl)methyl)pyrimidine-2,4(1H,3H)-diones (10a–d) (Series II), were synthesized using catalyst-free condensation of acid hydrazide 4 with different aromatic aldehydes and nucleophilic substitution reactions of chloromethylpyrimidine with different aliphatic amines , respectively. Compared to gefitinib (IC50 = 4.1 ± 0.01 μM) and thalidomide (IC50 = 13.4 ± 0.5 μM), compounds 6c and 10b exhibited moderate anti-proliferative activity against the MCF-7 breast cancer cell line, with IC50 values of 37.7 ± 3.6 and 31.8 ± 2.0 μM, respectively. They demonstrated selective cytotoxicity toward MCF-7 cells over normal WI38 fibroblasts, with IC50 values of 87.3 ± 2.6 μM (6c) and >100 μM (10b). Both compounds showed potent in vitro EGFR-TK enzyme inhibition, with IC50 values of 0.9 ± 0.03 μM (6c) and 0.7 ± 0.02 μM (10b). Molecular docking simulations revealed that 6c and 10b effectively bind to the EGFR ATP-binding site, forming hydrogen bonds with the key hinge region amino acid Met793, similar to gefitinib. In silico studies confirmed that both compounds fit the criteria for orally bioavailable drug candidates, showing high gastrointestinal absorption and no predicted adverse effects on the central nervous system or liver. Additionally, both compounds were predicted to be non-carcinogenic and non-mutagenic.
期刊介绍:
An international, peer-reviewed journal covering all of the chemical sciences, including multidisciplinary and emerging areas. RSC Advances is a gold open access journal allowing researchers free access to research articles, and offering an affordable open access publishing option for authors around the world.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


