Vishnu Raghuraman, Archana Verma, Nicholas E Jackson
{"title":"有机半导体中分子掺杂的全原子反应蒙特卡罗分子动力学。","authors":"Vishnu Raghuraman, Archana Verma, Nicholas E Jackson","doi":"10.1021/acs.jctc.5c01206","DOIUrl":null,"url":null,"abstract":"<p><p>The computational study of molecular doping in organic semiconductors (OSCs) is challenged by multiple competing length and time scales. We present an all-atom Reactive Monte Carlo Molecular Dynamics (RMCMD) method for quantitatively determining molecular doping efficiency in OSCs. A Metropolis criterion is employed for the doping reaction, which is parametrized from density functional theory (DFT) calculations of energetics and atomic partial charges of the doped and neutral molecular species. Polaronic effects are included in the RMCMD method to enable geometric reorganization upon doping, including the flattening of the inter-ring dihedrals. Extensions to partial charge transfer states are also developed to allow for the inclusion of charge transfer complexes. To demonstrate the validity of the approach, the doping efficiency and radial distribution function of a poly(3-hexylthiophene) system doped with 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane are calculated in an amorphous morphology. The method is implemented as a fix in LAMMPS, with the code made publicly available on GitHub.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2025-10-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"All-Atom Reactive Monte Carlo Molecular Dynamics for Molecular Doping in Organic Semiconductors.\",\"authors\":\"Vishnu Raghuraman, Archana Verma, Nicholas E Jackson\",\"doi\":\"10.1021/acs.jctc.5c01206\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The computational study of molecular doping in organic semiconductors (OSCs) is challenged by multiple competing length and time scales. We present an all-atom Reactive Monte Carlo Molecular Dynamics (RMCMD) method for quantitatively determining molecular doping efficiency in OSCs. A Metropolis criterion is employed for the doping reaction, which is parametrized from density functional theory (DFT) calculations of energetics and atomic partial charges of the doped and neutral molecular species. Polaronic effects are included in the RMCMD method to enable geometric reorganization upon doping, including the flattening of the inter-ring dihedrals. Extensions to partial charge transfer states are also developed to allow for the inclusion of charge transfer complexes. To demonstrate the validity of the approach, the doping efficiency and radial distribution function of a poly(3-hexylthiophene) system doped with 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane are calculated in an amorphous morphology. The method is implemented as a fix in LAMMPS, with the code made publicly available on GitHub.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-10-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.5c01206\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.5c01206","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
All-Atom Reactive Monte Carlo Molecular Dynamics for Molecular Doping in Organic Semiconductors.
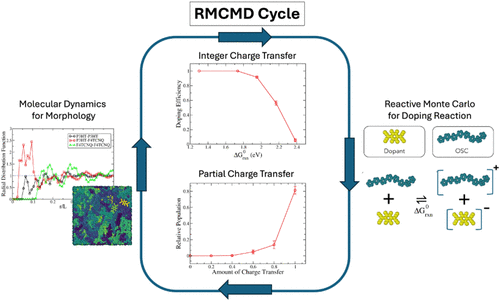
The computational study of molecular doping in organic semiconductors (OSCs) is challenged by multiple competing length and time scales. We present an all-atom Reactive Monte Carlo Molecular Dynamics (RMCMD) method for quantitatively determining molecular doping efficiency in OSCs. A Metropolis criterion is employed for the doping reaction, which is parametrized from density functional theory (DFT) calculations of energetics and atomic partial charges of the doped and neutral molecular species. Polaronic effects are included in the RMCMD method to enable geometric reorganization upon doping, including the flattening of the inter-ring dihedrals. Extensions to partial charge transfer states are also developed to allow for the inclusion of charge transfer complexes. To demonstrate the validity of the approach, the doping efficiency and radial distribution function of a poly(3-hexylthiophene) system doped with 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane are calculated in an amorphous morphology. The method is implemented as a fix in LAMMPS, with the code made publicly available on GitHub.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


