在单细胞RNA-Seq数据集中创建合成细胞的混合关注专家嵌入基于流的生成模型。
摘要
在细胞水平上进行的单细胞RNA-seq (scRNAseq)分析旨在了解组织切片的细胞景观,提供对罕见细胞类型的见解,并鉴定用于注释不同细胞类型的标记基因。ScRNAseq分析被广泛应用于癌症研究,以了解肿瘤异质性、疾病进展和对治疗的耐药性。单细胞数据处理是一项具有挑战性的任务,因为它具有高维性、稀疏性和不平衡的类(细胞类型)分布。准确的细胞类型鉴定高度依赖于预处理和质量控制步骤。为了解决这些问题,生成模型近年来得到了广泛的应用。常用的技术包括变分自编码器(VAE)、生成对抗网络(gan)、基于高斯的方法,以及最近的基于流的生成模型。我们开发了一个掩模仿射自回归变换嵌入FB (MAF-FB)模型。然后,为了进一步改进MAF-FB,我们在其上加入了注意机制的混合专家(MOE),从而得到了我们提出的MOE- fb模型。我们对基本生成模型进行了比较分析,旨在为开发新型自动化scRNAseq数据分析系统提供初步指导。我们通过结合来自胰腺组织切片的四个数据集进行了大规模分析,为了进一步的泛化评估,我们使用了外周血单个核细胞(PBMC68K和PBMC3K)和人类细胞图谱骨髓(HCA-BM10K)数据集。我们使用VAE、GAN、Gaussian Copula和Automated cell - type informed Introspective Variational Autoencoder (ACTIVA),并将它们与我们的两种新型FB模型MAF-FB和MOE-FB进行了ScRnaseq合成的比较。为了评估生成模型的性能,我们使用了各种差异度量并执行了自动细胞类型分类任务。我们还鉴定了每种细胞类型的差异表达基因,并推断了基于不同细胞类型对的配体-受体结合的细胞-细胞相互作用。在生成模型中,FB模型,尤其是MOE-FB,在所有实验设置中,与基线测试集和细胞类型分类任务相比,在差异指标上始终优于其他模型(f1得分为0.90,精度为0.89,召回率为0.92)。MOE-FB产生了生物学上更相关的合成数据,从合成细胞推断出的基于配体受体的细胞-细胞相互作用与原始数据非常相似,相对于相应的胰腺测试集,RMSE为0.65。这些发现突出了FB模型,特别是MOE-FB在scRNAseq分析中的潜力和前景。
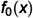
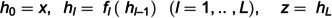
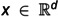
Single-cell RNA-seq (scRNAseq) analyses performed at the cellular level aim to understand the cellular landscape of tissue sections, offer insights into rare cell-types, and identify marker genes for annotating distinct cell types. ScRNAseq analyses are widely applied to cancer research to understand tumor heterogeneity, disease progression, and resistance to therapy. Single-cell data processing is a challenging task due to its high-dimensionality, sparsity, and having imbalanced class(cell-type) distributions. An accurate cell-type identification is highly dependent on preprocessing and quality control steps. To address these issues, generative models have been widely used in recent years. Techniques frequently used include Variational Autoencoders (VAE), Generative Adversarial Networks (GANs), Gaussian-based methods, and, more recently, Flow-based (FB) generative models. We developed a Masked Affine Autoregressive transform-embedded FB (MAF-FB) model. Then, to improve MAF-FB further, we incorporated a mixture of experts (MOE) of attention mechanisms on top of it, resulting in our proposed MOE-FB model. We conducted a comparative analysis of fundamental generative models, aiming to serve as a preliminary guidance for developing novel automated scRNAseq data analysis systems. We performed a large-scale analysis by combiningfour datasets derived from pancreatic tissue sections and for further generalizability assessments, we employed Peripheral Blood Mononuclear Cells (PBMC68K and PBMC3K) and Human Cell Atlas Bone Marrow (HCA-BM10K) datasets. We utilized VAE, GAN, Gaussian Copula, and Automated Cell-Type-informed Introspective Variational Autoencoder (ACTIVA), and compared them against our two novel FB models, MAF-FB and MOE-FB for ScRnaseq synthesis. To evaluate the performances of generative models, we used various discrepancy metrics and performed automated cell-type classification tasks. We also identified differentially expressed genes for each cell type, and inferred cell-cell interactions based on ligand-receptor bindings across distinct cell-type pairs. Among the generative models, FB models, especially MOE-FB, consistently outperformed others across all experimental setups in both discrepancy metrics with comparison to the baseline test set and cell-type classification tasks (with an F1-score of 0.90 precision of 0.89 and recall of 0.92 for the integrated pancreatic datasets). MOE-FB produced biologically more relevant synthetic data, and ligand-receptor-based cell-cell interactions inferred from the synthetic cells closely resemble the original data, achieving an RMSE of 0.65 against the corresponding pancreatic test set. These findings highlight the potential and promising use of FB models, especially MOE-FB, in scRNAseq analyses.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


