{"title":"计算电化学中隐式溶剂化的高斯周期大正则密度泛函理论。","authors":"Anton Z Ni, Adam Rettig, Joonho Lee","doi":"10.1021/acs.jctc.5c01403","DOIUrl":null,"url":null,"abstract":"<p><p>We present a numerical method for grand canonical density functional theory (DFT) tailored to solid-state systems, employing Gaussian-type orbitals as the primary basis. Our approach directly minimizes the grand canonical free energy using the density matrix as the sole variational parameter, while self-consistently updating the electron number between self-consistent field iterations. To enable realistic electrochemical modeling, we integrate this approach with implicit solvation models. Our solvation scheme introduces less than 50% overhead relative to gas-phase calculations. Compared to existing plane wave-based implementations, our method shows improved robustness in grand canonical simulations. We validate the approach by modeling corrosion at silver surfaces, finding excellent agreement with previous studies. Our method is implemented in the quantum chemistry software Q-Chem. This work lays the groundwork for future wave function-based simulations beyond DFT under electrochemical operando conditions.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2025-10-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Gaussian-Based Periodic Grand Canonical Density Functional Theory with Implicit Solvation for Computational Electrochemistry.\",\"authors\":\"Anton Z Ni, Adam Rettig, Joonho Lee\",\"doi\":\"10.1021/acs.jctc.5c01403\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>We present a numerical method for grand canonical density functional theory (DFT) tailored to solid-state systems, employing Gaussian-type orbitals as the primary basis. Our approach directly minimizes the grand canonical free energy using the density matrix as the sole variational parameter, while self-consistently updating the electron number between self-consistent field iterations. To enable realistic electrochemical modeling, we integrate this approach with implicit solvation models. Our solvation scheme introduces less than 50% overhead relative to gas-phase calculations. Compared to existing plane wave-based implementations, our method shows improved robustness in grand canonical simulations. We validate the approach by modeling corrosion at silver surfaces, finding excellent agreement with previous studies. Our method is implemented in the quantum chemistry software Q-Chem. This work lays the groundwork for future wave function-based simulations beyond DFT under electrochemical operando conditions.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-10-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.5c01403\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.5c01403","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Gaussian-Based Periodic Grand Canonical Density Functional Theory with Implicit Solvation for Computational Electrochemistry.
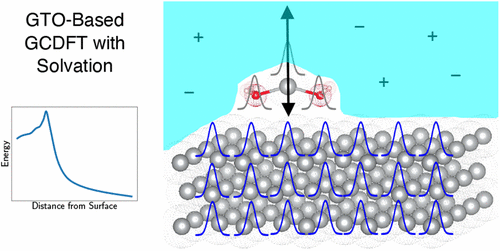
We present a numerical method for grand canonical density functional theory (DFT) tailored to solid-state systems, employing Gaussian-type orbitals as the primary basis. Our approach directly minimizes the grand canonical free energy using the density matrix as the sole variational parameter, while self-consistently updating the electron number between self-consistent field iterations. To enable realistic electrochemical modeling, we integrate this approach with implicit solvation models. Our solvation scheme introduces less than 50% overhead relative to gas-phase calculations. Compared to existing plane wave-based implementations, our method shows improved robustness in grand canonical simulations. We validate the approach by modeling corrosion at silver surfaces, finding excellent agreement with previous studies. Our method is implemented in the quantum chemistry software Q-Chem. This work lays the groundwork for future wave function-based simulations beyond DFT under electrochemical operando conditions.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


