Kerri Spontarelli Fruit, J Fernando Olivera, Nicolas Colmano, Shawn J Bird, Brett A McCray, Sho T Yano, Steven S Scherer, Pablo Artigas
{"title":"复发性ATP1 A1变异p.Gly549Arg与中间CMT和Na, k - atp酶功能丧失的关系","authors":"Kerri Spontarelli Fruit, J Fernando Olivera, Nicolas Colmano, Shawn J Bird, Brett A McCray, Sho T Yano, Steven S Scherer, Pablo Artigas","doi":"10.1212/NXG.0000000000200309","DOIUrl":null,"url":null,"abstract":"<p><strong>Background and objectives: </strong>Charcot-Marie-Tooth (CMT) disease comprises a group of inherited peripheral neuropathies caused by pathogenic variants in various genes, including <i>ATP1A1</i>. This gene encodes the ubiquitous α1 subunit of the sodium pump that generates the Na<sup>+</sup> and K<sup>+</sup> gradients that are essential for neuronal survival and excitability. We present the clinical cases of 2 unrelated patients with the same <i>ATP1A1</i> variant causing dominant intermediate CMT disease and the functional characterization of the variant in the heterologous expression system.</p><p><strong>Methods: </strong>The patients were evaluated by clinical EMG and by whole-exome sequencing. The function of sodium pump variants was studied with voltage clamp electrophysiology or using ouabain survival curves after heterologous expression in <i>Xenopus</i> oocytes or HEK293 cells, respectively. Localization of the variants was evaluated by fluorescence microscopy of HEK293 cells expressing fluorescently tagged sodium pumps.</p><p><strong>Results: </strong>We describe the cases of 2 unrelated patients who presented in their second decade with a length-dependent and slowly progressive intermediate neuropathy with both axonal and demyelinating features. Whole-exome sequencing identified a de novo c.1645G>A heterozygous variant in <i>ATP1A1</i> (p.Gly549Arg) in both patients. The pathogenic nature of the variant was tested through a detailed evaluation of the functional consequences of the Gly549Arg substitution using 2 heterologous expression systems and functional assays that included survival curves of transfected cells and electrophysiology. Patch clamp and 2-electrode voltage clamp electrophysiology experiments showed that the Gly549Arg variant reduced NKA function (≥50%), mainly due to a lower NKA density at the plasma membrane and, to a lesser extent, due to a reduced apparent affinity for intracellular Na<sup>+</sup>. The reduced plasma membrane density was also observed in HEK293 cells simultaneously expressing wildtype and Gly549Arg variants, marked with fluorescent proteins of different colors, suggesting that the mutant may be partially retained in intracellular membranes. No clear dominant-negative effects were identified in these experimental systems.</p><p><strong>Discussion: </strong>Our results demonstrate that the pathogenic nature of this variant causes considerable loss of function due to diminished plasma membrane localization and kinetic impairments on the enzyme, without obvious dominant-negative effects. Our findings are similar to those previously reported for other CMT disease-causing ATP1A1 variants.</p>","PeriodicalId":48613,"journal":{"name":"Neurology-Genetics","volume":"11 5","pages":"e200309"},"PeriodicalIF":3.7000,"publicationDate":"2025-09-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12488841/pdf/","citationCount":"0","resultStr":"{\"title\":\"Association of the Recurrent <i>ATP1</i> <i>A1</i> Variant p.Gly549Arg With Intermediate CMT and Loss of Na,K-ATPase Function.\",\"authors\":\"Kerri Spontarelli Fruit, J Fernando Olivera, Nicolas Colmano, Shawn J Bird, Brett A McCray, Sho T Yano, Steven S Scherer, Pablo Artigas\",\"doi\":\"10.1212/NXG.0000000000200309\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background and objectives: </strong>Charcot-Marie-Tooth (CMT) disease comprises a group of inherited peripheral neuropathies caused by pathogenic variants in various genes, including <i>ATP1A1</i>. This gene encodes the ubiquitous α1 subunit of the sodium pump that generates the Na<sup>+</sup> and K<sup>+</sup> gradients that are essential for neuronal survival and excitability. We present the clinical cases of 2 unrelated patients with the same <i>ATP1A1</i> variant causing dominant intermediate CMT disease and the functional characterization of the variant in the heterologous expression system.</p><p><strong>Methods: </strong>The patients were evaluated by clinical EMG and by whole-exome sequencing. The function of sodium pump variants was studied with voltage clamp electrophysiology or using ouabain survival curves after heterologous expression in <i>Xenopus</i> oocytes or HEK293 cells, respectively. Localization of the variants was evaluated by fluorescence microscopy of HEK293 cells expressing fluorescently tagged sodium pumps.</p><p><strong>Results: </strong>We describe the cases of 2 unrelated patients who presented in their second decade with a length-dependent and slowly progressive intermediate neuropathy with both axonal and demyelinating features. Whole-exome sequencing identified a de novo c.1645G>A heterozygous variant in <i>ATP1A1</i> (p.Gly549Arg) in both patients. The pathogenic nature of the variant was tested through a detailed evaluation of the functional consequences of the Gly549Arg substitution using 2 heterologous expression systems and functional assays that included survival curves of transfected cells and electrophysiology. Patch clamp and 2-electrode voltage clamp electrophysiology experiments showed that the Gly549Arg variant reduced NKA function (≥50%), mainly due to a lower NKA density at the plasma membrane and, to a lesser extent, due to a reduced apparent affinity for intracellular Na<sup>+</sup>. The reduced plasma membrane density was also observed in HEK293 cells simultaneously expressing wildtype and Gly549Arg variants, marked with fluorescent proteins of different colors, suggesting that the mutant may be partially retained in intracellular membranes. No clear dominant-negative effects were identified in these experimental systems.</p><p><strong>Discussion: </strong>Our results demonstrate that the pathogenic nature of this variant causes considerable loss of function due to diminished plasma membrane localization and kinetic impairments on the enzyme, without obvious dominant-negative effects. Our findings are similar to those previously reported for other CMT disease-causing ATP1A1 variants.</p>\",\"PeriodicalId\":48613,\"journal\":{\"name\":\"Neurology-Genetics\",\"volume\":\"11 5\",\"pages\":\"e200309\"},\"PeriodicalIF\":3.7000,\"publicationDate\":\"2025-09-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12488841/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurology-Genetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1212/NXG.0000000000200309\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/10/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurology-Genetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1212/NXG.0000000000200309","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/10/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Association of the Recurrent ATP1A1 Variant p.Gly549Arg With Intermediate CMT and Loss of Na,K-ATPase Function.
Background and objectives: Charcot-Marie-Tooth (CMT) disease comprises a group of inherited peripheral neuropathies caused by pathogenic variants in various genes, including ATP1A1. This gene encodes the ubiquitous α1 subunit of the sodium pump that generates the Na+ and K+ gradients that are essential for neuronal survival and excitability. We present the clinical cases of 2 unrelated patients with the same ATP1A1 variant causing dominant intermediate CMT disease and the functional characterization of the variant in the heterologous expression system.
Methods: The patients were evaluated by clinical EMG and by whole-exome sequencing. The function of sodium pump variants was studied with voltage clamp electrophysiology or using ouabain survival curves after heterologous expression in Xenopus oocytes or HEK293 cells, respectively. Localization of the variants was evaluated by fluorescence microscopy of HEK293 cells expressing fluorescently tagged sodium pumps.
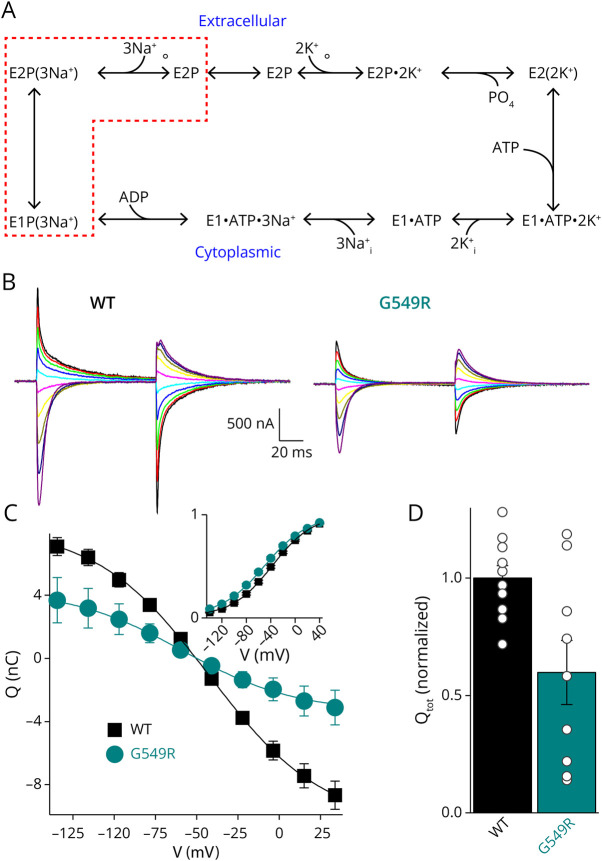
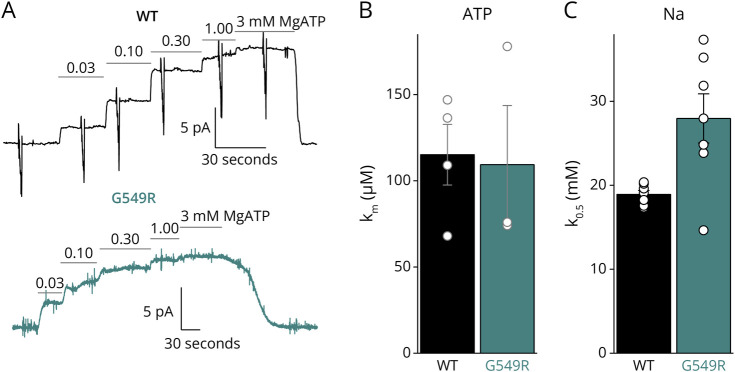
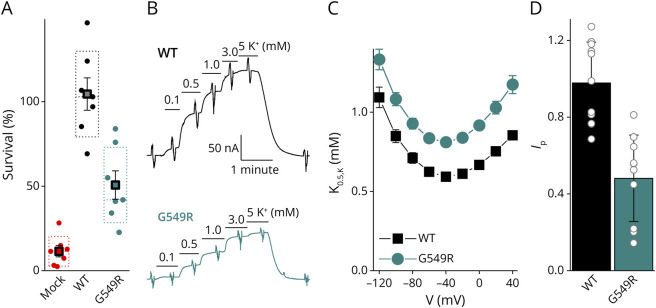
Results: We describe the cases of 2 unrelated patients who presented in their second decade with a length-dependent and slowly progressive intermediate neuropathy with both axonal and demyelinating features. Whole-exome sequencing identified a de novo c.1645G>A heterozygous variant in ATP1A1 (p.Gly549Arg) in both patients. The pathogenic nature of the variant was tested through a detailed evaluation of the functional consequences of the Gly549Arg substitution using 2 heterologous expression systems and functional assays that included survival curves of transfected cells and electrophysiology. Patch clamp and 2-electrode voltage clamp electrophysiology experiments showed that the Gly549Arg variant reduced NKA function (≥50%), mainly due to a lower NKA density at the plasma membrane and, to a lesser extent, due to a reduced apparent affinity for intracellular Na+. The reduced plasma membrane density was also observed in HEK293 cells simultaneously expressing wildtype and Gly549Arg variants, marked with fluorescent proteins of different colors, suggesting that the mutant may be partially retained in intracellular membranes. No clear dominant-negative effects were identified in these experimental systems.
Discussion: Our results demonstrate that the pathogenic nature of this variant causes considerable loss of function due to diminished plasma membrane localization and kinetic impairments on the enzyme, without obvious dominant-negative effects. Our findings are similar to those previously reported for other CMT disease-causing ATP1A1 variants.
期刊介绍:
Neurology: Genetics is an online open access journal publishing peer-reviewed reports in the field of neurogenetics. Original articles in all areas of neurogenetics will be published including rare and common genetic variation, genotype-phenotype correlations, outlier phenotypes as a result of mutations in known disease-genes, and genetic variations with a putative link to diseases. This will include studies reporting on genetic disease risk and pharmacogenomics. In addition, Neurology: Genetics will publish results of gene-based clinical trials (viral, ASO, etc.). Genetically engineered model systems are not a primary focus of Neurology: Genetics, but studies using model systems for treatment trials are welcome, including well-powered studies reporting negative results.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


