Pontus Svensson*, , , Fotios Kalkavouras, , , Uwe Hernandez Acosta, , , Zhandos A. Moldabekov, , , Panagiotis Tolias, , , Jan Vorberger, , and , Tobias Dornheim,
{"title":"加速自由能估计在从头算路径积分蒙特卡罗模拟","authors":"Pontus Svensson*, , , Fotios Kalkavouras, , , Uwe Hernandez Acosta, , , Zhandos A. Moldabekov, , , Panagiotis Tolias, , , Jan Vorberger, , and , Tobias Dornheim, ","doi":"10.1021/acs.jpclett.5c02193","DOIUrl":null,"url":null,"abstract":"<p >We present a methodology for accelerating the estimation of the free energy from path integral Monte Carlo simulations by considering an intermediate artificial reference system where interactions are inexpensive to evaluate numerically. Using the spherically averaged Ewald interaction as this intermediate reference system for the uniform electron gas, the interaction contribution for the free energy was evaluated up to 18 times faster than the Ewald-only method. Furthermore, an extrapolation technique with respect to the quantum statistics was tested and applied to alleviate the Fermion sign problem. Combining these two techniques enabled the evaluation of the free energy for a system of 1000 electrons, where both finite-size and statistical errors are below chemical accuracy. The general procedure can be applied to systems relevant for planetary and inertial confinement fusion modeling with low to moderate levels of quantum degeneracy.</p>","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"16 41","pages":"10639–10646"},"PeriodicalIF":4.6000,"publicationDate":"2025-10-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/pdf/10.1021/acs.jpclett.5c02193","citationCount":"0","resultStr":"{\"title\":\"Accelerated Free Energy Estimation in Ab Initio Path Integral Monte Carlo Simulations\",\"authors\":\"Pontus Svensson*, , , Fotios Kalkavouras, , , Uwe Hernandez Acosta, , , Zhandos A. Moldabekov, , , Panagiotis Tolias, , , Jan Vorberger, , and , Tobias Dornheim, \",\"doi\":\"10.1021/acs.jpclett.5c02193\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We present a methodology for accelerating the estimation of the free energy from path integral Monte Carlo simulations by considering an intermediate artificial reference system where interactions are inexpensive to evaluate numerically. Using the spherically averaged Ewald interaction as this intermediate reference system for the uniform electron gas, the interaction contribution for the free energy was evaluated up to 18 times faster than the Ewald-only method. Furthermore, an extrapolation technique with respect to the quantum statistics was tested and applied to alleviate the Fermion sign problem. Combining these two techniques enabled the evaluation of the free energy for a system of 1000 electrons, where both finite-size and statistical errors are below chemical accuracy. The general procedure can be applied to systems relevant for planetary and inertial confinement fusion modeling with low to moderate levels of quantum degeneracy.</p>\",\"PeriodicalId\":62,\"journal\":{\"name\":\"The Journal of Physical Chemistry Letters\",\"volume\":\"16 41\",\"pages\":\"10639–10646\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2025-10-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/pdf/10.1021/acs.jpclett.5c02193\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry Letters\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpclett.5c02193\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpclett.5c02193","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Accelerated Free Energy Estimation in Ab Initio Path Integral Monte Carlo Simulations
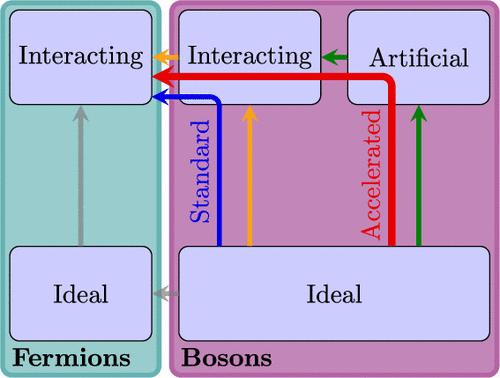
We present a methodology for accelerating the estimation of the free energy from path integral Monte Carlo simulations by considering an intermediate artificial reference system where interactions are inexpensive to evaluate numerically. Using the spherically averaged Ewald interaction as this intermediate reference system for the uniform electron gas, the interaction contribution for the free energy was evaluated up to 18 times faster than the Ewald-only method. Furthermore, an extrapolation technique with respect to the quantum statistics was tested and applied to alleviate the Fermion sign problem. Combining these two techniques enabled the evaluation of the free energy for a system of 1000 electrons, where both finite-size and statistical errors are below chemical accuracy. The general procedure can be applied to systems relevant for planetary and inertial confinement fusion modeling with low to moderate levels of quantum degeneracy.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


