{"title":"基于数据驱动的几何图形神经网络的端到端反应场能量建模。","authors":"Yongxian Wu, , , Qiang Zhu, , and , Ray Luo*, ","doi":"10.1021/acs.jctc.5c01193","DOIUrl":null,"url":null,"abstract":"<p >Electrostatic interactions are fundamental to the structure, dynamics, and function of biomolecules, with broad applications in protein–ligand binding, enzymatic catalysis, and nucleic acid regulation. The Poisson–Boltzmann (PB) equation provides a physically grounded framework for modeling these interactions. However, solving the PB equation for large and complex biomolecular systems remains computationally expensive as traditional numerical solvers scale poorly with system size. While the Generalized Born (GB) model offers a more computationally efficient approximation, it does so at the cost of reduced accuracy relative to full PB solutions. To overcome these limitations, we propose PBGNN, a novel end-to-end framework that uses data-driven geometric graph neural networks to directly approximate PB electrostatic energies without relying on the GB approximation. PBGNN incorporates sinusoidal embeddings of atomic charges and a message-passing architecture to efficiently capture long-range interactions in large biomolecules. To address training instability caused by high variance in atomic electrostatic potentials, we introduce a charge-weighted mean squared error (CMSE) optimization objective that improves convergence. We benchmark PBGNN on the AMBER PBSA suite and PBSMALL, a new dataset designed for rapid evaluation of small-molecule electrostatics in drug discovery contexts. The results demonstrate that PBGNN consistently achieves high accuracy in predicting the PB energy with linear computational complexity. Furthermore, it provides reliable and precise PB free energy predictions for both large biomolecular complexes and small-molecule datasets, showcasing its strong generalizability, scalability, and potential utility in drug discovery tasks requiring accurate electrostatic modeling of small molecules. Comprehensive ablation studies further reveal the impact of architectural components, such as geometric representation, objective design, and cutoff strategy, informing future research directions. Finally, we release PBGNN as an open-source, self-contained codebase, along with preprocessed datasets and a complete training and evaluation pipeline, to support scalable and accurate electrostatic analysis that facilitates future research in associated areas.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9710–9725"},"PeriodicalIF":5.5000,"publicationDate":"2025-10-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"End-to-End Modeling of Reaction Field Energy Using Data-Driven Geometric Graph Neural Networks\",\"authors\":\"Yongxian Wu, , , Qiang Zhu, , and , Ray Luo*, \",\"doi\":\"10.1021/acs.jctc.5c01193\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Electrostatic interactions are fundamental to the structure, dynamics, and function of biomolecules, with broad applications in protein–ligand binding, enzymatic catalysis, and nucleic acid regulation. The Poisson–Boltzmann (PB) equation provides a physically grounded framework for modeling these interactions. However, solving the PB equation for large and complex biomolecular systems remains computationally expensive as traditional numerical solvers scale poorly with system size. While the Generalized Born (GB) model offers a more computationally efficient approximation, it does so at the cost of reduced accuracy relative to full PB solutions. To overcome these limitations, we propose PBGNN, a novel end-to-end framework that uses data-driven geometric graph neural networks to directly approximate PB electrostatic energies without relying on the GB approximation. PBGNN incorporates sinusoidal embeddings of atomic charges and a message-passing architecture to efficiently capture long-range interactions in large biomolecules. To address training instability caused by high variance in atomic electrostatic potentials, we introduce a charge-weighted mean squared error (CMSE) optimization objective that improves convergence. We benchmark PBGNN on the AMBER PBSA suite and PBSMALL, a new dataset designed for rapid evaluation of small-molecule electrostatics in drug discovery contexts. The results demonstrate that PBGNN consistently achieves high accuracy in predicting the PB energy with linear computational complexity. Furthermore, it provides reliable and precise PB free energy predictions for both large biomolecular complexes and small-molecule datasets, showcasing its strong generalizability, scalability, and potential utility in drug discovery tasks requiring accurate electrostatic modeling of small molecules. Comprehensive ablation studies further reveal the impact of architectural components, such as geometric representation, objective design, and cutoff strategy, informing future research directions. Finally, we release PBGNN as an open-source, self-contained codebase, along with preprocessed datasets and a complete training and evaluation pipeline, to support scalable and accurate electrostatic analysis that facilitates future research in associated areas.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"21 19\",\"pages\":\"9710–9725\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-10-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01193\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01193","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
静电相互作用是生物分子结构、动力学和功能的基础,在蛋白质-配体结合、酶催化和核酸调节等领域有着广泛的应用。泊松-玻尔兹曼(PB)方程为这些相互作用的建模提供了一个物理基础框架。然而,求解大型复杂生物分子系统的PB方程仍然是计算上昂贵的,因为传统的数值求解方法与系统规模的比例很低。虽然Generalized Born (GB)模型提供了一个计算效率更高的近似值,但相对于完整的PB解决方案,它的代价是降低了精度。为了克服这些限制,我们提出了PBGNN,这是一种新颖的端到端框架,它使用数据驱动的几何图形神经网络直接近似PB静电能量,而不依赖于GB近似。PBGNN结合了原子电荷的正弦嵌入和信息传递架构,以有效地捕获大型生物分子中的远程相互作用。为了解决由原子静电电位的高方差引起的训练不稳定性,我们引入了电荷加权均方误差(CMSE)优化目标,以提高收敛性。我们在AMBER PBSA套件和PBSMALL上对PBGNN进行基准测试,PBSMALL是一个新的数据集,旨在快速评估药物发现环境中的小分子静电。结果表明,PBGNN在线性计算复杂度的PB能量预测中始终保持较高的精度。此外,它为大型生物分子复合物和小分子数据集提供了可靠和精确的PB自由能预测,展示了其强大的通用性,可扩展性,以及在需要精确的小分子静电建模的药物发现任务中的潜在实用性。综合消融研究进一步揭示了建筑构件的影响,如几何表征、客观设计和切断策略,为未来的研究方向提供信息。最后,我们将PBGNN作为一个开源的、自包含的代码库,以及预处理数据集和完整的培训和评估管道发布,以支持可扩展和准确的静电分析,从而促进相关领域的未来研究。
End-to-End Modeling of Reaction Field Energy Using Data-Driven Geometric Graph Neural Networks
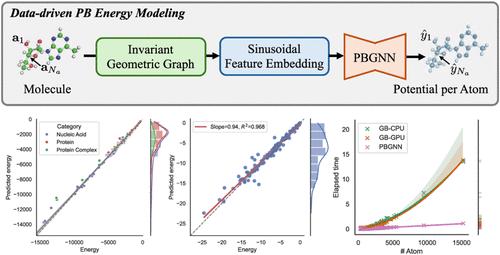
Electrostatic interactions are fundamental to the structure, dynamics, and function of biomolecules, with broad applications in protein–ligand binding, enzymatic catalysis, and nucleic acid regulation. The Poisson–Boltzmann (PB) equation provides a physically grounded framework for modeling these interactions. However, solving the PB equation for large and complex biomolecular systems remains computationally expensive as traditional numerical solvers scale poorly with system size. While the Generalized Born (GB) model offers a more computationally efficient approximation, it does so at the cost of reduced accuracy relative to full PB solutions. To overcome these limitations, we propose PBGNN, a novel end-to-end framework that uses data-driven geometric graph neural networks to directly approximate PB electrostatic energies without relying on the GB approximation. PBGNN incorporates sinusoidal embeddings of atomic charges and a message-passing architecture to efficiently capture long-range interactions in large biomolecules. To address training instability caused by high variance in atomic electrostatic potentials, we introduce a charge-weighted mean squared error (CMSE) optimization objective that improves convergence. We benchmark PBGNN on the AMBER PBSA suite and PBSMALL, a new dataset designed for rapid evaluation of small-molecule electrostatics in drug discovery contexts. The results demonstrate that PBGNN consistently achieves high accuracy in predicting the PB energy with linear computational complexity. Furthermore, it provides reliable and precise PB free energy predictions for both large biomolecular complexes and small-molecule datasets, showcasing its strong generalizability, scalability, and potential utility in drug discovery tasks requiring accurate electrostatic modeling of small molecules. Comprehensive ablation studies further reveal the impact of architectural components, such as geometric representation, objective design, and cutoff strategy, informing future research directions. Finally, we release PBGNN as an open-source, self-contained codebase, along with preprocessed datasets and a complete training and evaluation pipeline, to support scalable and accurate electrostatic analysis that facilitates future research in associated areas.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


