Damla Baysal Bakır, Özge Atay, Halime Yağmur, Gizem Kabadayı, Mehmet Kocabey, Suna Asilsoy, Nevin Uzuner
{"title":"扩大儿童共济失调毛细血管扩张症的临床频谱:一系列新的遗传变异,寻常性狼疮和超igm表型的病例。","authors":"Damla Baysal Bakır, Özge Atay, Halime Yağmur, Gizem Kabadayı, Mehmet Kocabey, Suna Asilsoy, Nevin Uzuner","doi":"10.1186/s13023-025-03942-7","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Ataxia-telangiectasia (A-T) is a rare autosomal recessive disorder caused by pathogenic ATM gene variants, characterised by progressive cerebellar ataxia, telangiectasia, immunodeficiency, and cancer predisposition. While its immunological and oncological complications are well-documented, clinical heterogeneity, particularly in cases with elevated IgM, poses diagnostic challenges.</p><p><strong>Methods: </strong>Following written informed consent, we retrospectively analysed four pediatric A-T patients followed in our clinic. Clinical, laboratory, and radiological data were reviewed, including immunoglobulin levels, vaccine antibody responses, lymphocyte subsets, and alpha-fetoprotein (AFP) levels. Diagnosis was established based on clinical and laboratory findings, supported by whole-exome sequencing (WES) and targeted ATM gene sequencing.</p><p><strong>Results: </strong>Our findings further support the association between the hyper-IgM phenotype and increased immune dysfunction in A-T. We report the first globally documented case of lupus vulgaris in an A-T patient and identify a previously unreported ATM variant in our country, expanding the disease spectrum. These findings highlight the need for further research on regional genetic variations and their clinical implications.</p><p><strong>Conclusion: </strong>This study highlights the importance of early diagnosis and genetic testing, particularly in atypical presentations. The recognition of novel infectious and autoimmune associations, along with novel variants, underscores the necessity of comprehensive, multidisciplinary follow-up and regional genetic screening efforts.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"500"},"PeriodicalIF":3.5000,"publicationDate":"2025-10-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12495667/pdf/","citationCount":"0","resultStr":"{\"title\":\"Expanding the clinical spectrum of pediatric ataxia-telangiectasia: a case series of novel genetic variants, lupus vulgaris, and hyper-IgM phenotypes.\",\"authors\":\"Damla Baysal Bakır, Özge Atay, Halime Yağmur, Gizem Kabadayı, Mehmet Kocabey, Suna Asilsoy, Nevin Uzuner\",\"doi\":\"10.1186/s13023-025-03942-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Ataxia-telangiectasia (A-T) is a rare autosomal recessive disorder caused by pathogenic ATM gene variants, characterised by progressive cerebellar ataxia, telangiectasia, immunodeficiency, and cancer predisposition. While its immunological and oncological complications are well-documented, clinical heterogeneity, particularly in cases with elevated IgM, poses diagnostic challenges.</p><p><strong>Methods: </strong>Following written informed consent, we retrospectively analysed four pediatric A-T patients followed in our clinic. Clinical, laboratory, and radiological data were reviewed, including immunoglobulin levels, vaccine antibody responses, lymphocyte subsets, and alpha-fetoprotein (AFP) levels. Diagnosis was established based on clinical and laboratory findings, supported by whole-exome sequencing (WES) and targeted ATM gene sequencing.</p><p><strong>Results: </strong>Our findings further support the association between the hyper-IgM phenotype and increased immune dysfunction in A-T. We report the first globally documented case of lupus vulgaris in an A-T patient and identify a previously unreported ATM variant in our country, expanding the disease spectrum. These findings highlight the need for further research on regional genetic variations and their clinical implications.</p><p><strong>Conclusion: </strong>This study highlights the importance of early diagnosis and genetic testing, particularly in atypical presentations. The recognition of novel infectious and autoimmune associations, along with novel variants, underscores the necessity of comprehensive, multidisciplinary follow-up and regional genetic screening efforts.</p>\",\"PeriodicalId\":19651,\"journal\":{\"name\":\"Orphanet Journal of Rare Diseases\",\"volume\":\"20 1\",\"pages\":\"500\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2025-10-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12495667/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Orphanet Journal of Rare Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13023-025-03942-7\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03942-7","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Expanding the clinical spectrum of pediatric ataxia-telangiectasia: a case series of novel genetic variants, lupus vulgaris, and hyper-IgM phenotypes.
Background: Ataxia-telangiectasia (A-T) is a rare autosomal recessive disorder caused by pathogenic ATM gene variants, characterised by progressive cerebellar ataxia, telangiectasia, immunodeficiency, and cancer predisposition. While its immunological and oncological complications are well-documented, clinical heterogeneity, particularly in cases with elevated IgM, poses diagnostic challenges.
Methods: Following written informed consent, we retrospectively analysed four pediatric A-T patients followed in our clinic. Clinical, laboratory, and radiological data were reviewed, including immunoglobulin levels, vaccine antibody responses, lymphocyte subsets, and alpha-fetoprotein (AFP) levels. Diagnosis was established based on clinical and laboratory findings, supported by whole-exome sequencing (WES) and targeted ATM gene sequencing.
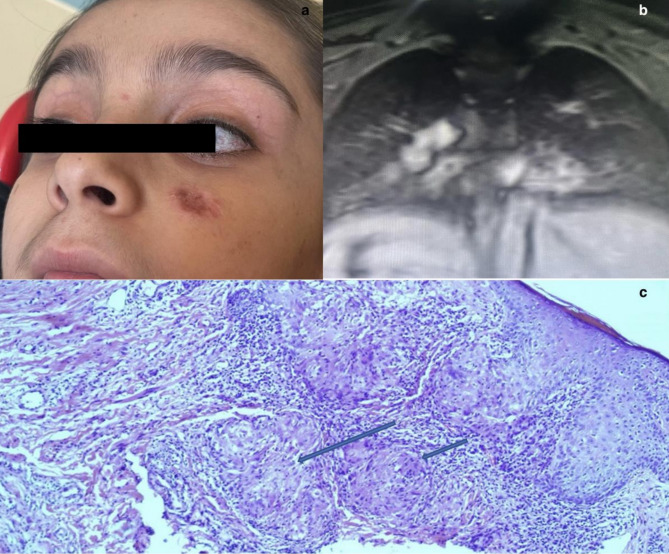
Results: Our findings further support the association between the hyper-IgM phenotype and increased immune dysfunction in A-T. We report the first globally documented case of lupus vulgaris in an A-T patient and identify a previously unreported ATM variant in our country, expanding the disease spectrum. These findings highlight the need for further research on regional genetic variations and their clinical implications.
Conclusion: This study highlights the importance of early diagnosis and genetic testing, particularly in atypical presentations. The recognition of novel infectious and autoimmune associations, along with novel variants, underscores the necessity of comprehensive, multidisciplinary follow-up and regional genetic screening efforts.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.



 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


