Yoosang Son, , , Yeseong Choi, , , Oleg V. Prezhdo*, , and , Hyungjun Kim*,
{"title":"利用热化高斯波包的半经典多态量子动力学。","authors":"Yoosang Son, , , Yeseong Choi, , , Oleg V. Prezhdo*, , and , Hyungjun Kim*, ","doi":"10.1021/acs.jctc.5c01001","DOIUrl":null,"url":null,"abstract":"<p >Electronic dynamics in condensed-phase systems are predominantly influenced by thermal effects and decoherence arising from the quantum bath coupled to the system. A system–bath Hamiltonian, where the ″system″ interacts with a ″bath″ of many harmonic oscillators, provides a foundational framework for investigating quantum dynamics with environmental effects. Here, we introduce a novel wave function-based method that enables real-time propagation of a finite-temperature harmonic bath wave function using classical equations of motion. We begin by purifying the thermal density matrix of a harmonic oscillator through the introduction of an auxiliary function, which corrects the decoherence behavior of standard Gaussian wavepackets. Using the path-integral formalism, we show that the resulting wavepacket evolves along classical trajectories under a sequence of shifted harmonic potentials. We then derive an explicit analytical form of this state, termed the thermalized Gaussian wavepacket (TGW). To propagate the TGW, we develop two numerical schemes; a stochastic hopping method for the finite coupling regime and a perturbative method tailored for the weak-coupling limit. Our simulations demonstrate that the TGW not only recovers Marcus theory rates in the weak-coupling limit for two-state systems, but also accurately reproduces numerically exact time-dependent Schrödinger equation (TDSE) or multiconfiguration time-dependent Hartree (MCTDH) results for two- and three-state models. These findings demonstrate that the density matrix evolution can be accurately simulated only by forward-propagating the TGW based on the classical trajectories. We envision that the TGW framework offers an efficient and accurate route to simulate electronic transition dynamics in real time, while rigorously incorporating both decoherence and thermal effects, making it a promising tool for investigating quantum dynamics in more complex and realistic systems.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9772–9783"},"PeriodicalIF":5.5000,"publicationDate":"2025-10-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Semiclassical Multistate Quantum Dynamics Using Thermalized Gaussian Wavepacket\",\"authors\":\"Yoosang Son, , , Yeseong Choi, , , Oleg V. Prezhdo*, , and , Hyungjun Kim*, \",\"doi\":\"10.1021/acs.jctc.5c01001\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Electronic dynamics in condensed-phase systems are predominantly influenced by thermal effects and decoherence arising from the quantum bath coupled to the system. A system–bath Hamiltonian, where the ″system″ interacts with a ″bath″ of many harmonic oscillators, provides a foundational framework for investigating quantum dynamics with environmental effects. Here, we introduce a novel wave function-based method that enables real-time propagation of a finite-temperature harmonic bath wave function using classical equations of motion. We begin by purifying the thermal density matrix of a harmonic oscillator through the introduction of an auxiliary function, which corrects the decoherence behavior of standard Gaussian wavepackets. Using the path-integral formalism, we show that the resulting wavepacket evolves along classical trajectories under a sequence of shifted harmonic potentials. We then derive an explicit analytical form of this state, termed the thermalized Gaussian wavepacket (TGW). To propagate the TGW, we develop two numerical schemes; a stochastic hopping method for the finite coupling regime and a perturbative method tailored for the weak-coupling limit. Our simulations demonstrate that the TGW not only recovers Marcus theory rates in the weak-coupling limit for two-state systems, but also accurately reproduces numerically exact time-dependent Schrödinger equation (TDSE) or multiconfiguration time-dependent Hartree (MCTDH) results for two- and three-state models. These findings demonstrate that the density matrix evolution can be accurately simulated only by forward-propagating the TGW based on the classical trajectories. We envision that the TGW framework offers an efficient and accurate route to simulate electronic transition dynamics in real time, while rigorously incorporating both decoherence and thermal effects, making it a promising tool for investigating quantum dynamics in more complex and realistic systems.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"21 19\",\"pages\":\"9772–9783\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-10-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01001\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01001","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
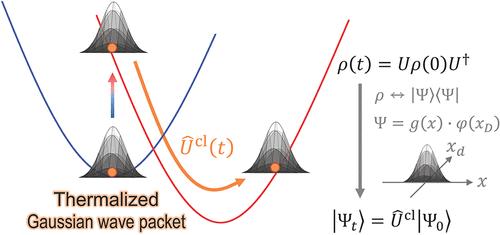
Semiclassical Multistate Quantum Dynamics Using Thermalized Gaussian Wavepacket
Electronic dynamics in condensed-phase systems are predominantly influenced by thermal effects and decoherence arising from the quantum bath coupled to the system. A system–bath Hamiltonian, where the ″system″ interacts with a ″bath″ of many harmonic oscillators, provides a foundational framework for investigating quantum dynamics with environmental effects. Here, we introduce a novel wave function-based method that enables real-time propagation of a finite-temperature harmonic bath wave function using classical equations of motion. We begin by purifying the thermal density matrix of a harmonic oscillator through the introduction of an auxiliary function, which corrects the decoherence behavior of standard Gaussian wavepackets. Using the path-integral formalism, we show that the resulting wavepacket evolves along classical trajectories under a sequence of shifted harmonic potentials. We then derive an explicit analytical form of this state, termed the thermalized Gaussian wavepacket (TGW). To propagate the TGW, we develop two numerical schemes; a stochastic hopping method for the finite coupling regime and a perturbative method tailored for the weak-coupling limit. Our simulations demonstrate that the TGW not only recovers Marcus theory rates in the weak-coupling limit for two-state systems, but also accurately reproduces numerically exact time-dependent Schrödinger equation (TDSE) or multiconfiguration time-dependent Hartree (MCTDH) results for two- and three-state models. These findings demonstrate that the density matrix evolution can be accurately simulated only by forward-propagating the TGW based on the classical trajectories. We envision that the TGW framework offers an efficient and accurate route to simulate electronic transition dynamics in real time, while rigorously incorporating both decoherence and thermal effects, making it a promising tool for investigating quantum dynamics in more complex and realistic systems.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


