{"title":"从系统发育数据确定接触者追踪对艾滋病毒流行病学推断的影响。","authors":"Michael D Kupperman, Ruian Ke, Thomas Leitner","doi":"10.1093/ve/veaf068","DOIUrl":null,"url":null,"abstract":"<p><p>Robust sampling methods are foundational to inferences using phylogenies. Yet the impact of using contact tracing, a type of non-uniform sampling used in public health applications such as infectious disease outbreak investigations, has not been investigated in the molecular epidemiology field. To understand how contact tracing influences a recovered phylogeny, we developed a new simulation tool called SEEPS (Sequence Evolution and Epidemiological Process Simulator) that allows for the simulation of contact tracing and the resulting transmission tree, pathogen phylogeny, and corresponding virus genetic sequences. Importantly, SEEPS takes within-host evolution into account when generating pathogen phylogenies and sequences from transmission histories. Using SEEPS, we demonstrate that contact tracing can significantly impact the structure of the resulting tree, as described by popular tree statistics. Contact tracing generates phylogenies that are less balanced than the underlying transmission process, less representative of the larger epidemiological process, and affects the internal/external branch length ratios that characterize specific epidemiological scenarios. We also examined real data from a 2007-2008 Swedish HIV-1 outbreak and the broader 1998-2010 European HIV-1 epidemic to highlight the differences in contact tracing and expected phylogenies. Aided by SEEPS, we show that the data collection of the Swedish outbreak was strongly influenced by contact tracing even after downsampling, while the broader European Union epidemic showed little evidence of universal contact tracing, agreeing with the known epidemiological information about sampling and spread. Overall, our results highlight the importance of including possible non-uniform sampling schemes when examining phylogenetic trees. For that, SEEPS serves as a useful tool to evaluate such impacts, thereby facilitating better phylogenetic inferences of the characteristics of a disease outbreak. SEEPS is available at https://github.com/MolEvolEpid/SEEPS.</p>","PeriodicalId":56026,"journal":{"name":"Virus Evolution","volume":"11 1","pages":"veaf068"},"PeriodicalIF":4.0000,"publicationDate":"2025-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12481701/pdf/","citationCount":"0","resultStr":"{\"title\":\"Identifying impacts of contact tracing on HIV epidemiological inference from phylogenetic data.\",\"authors\":\"Michael D Kupperman, Ruian Ke, Thomas Leitner\",\"doi\":\"10.1093/ve/veaf068\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Robust sampling methods are foundational to inferences using phylogenies. Yet the impact of using contact tracing, a type of non-uniform sampling used in public health applications such as infectious disease outbreak investigations, has not been investigated in the molecular epidemiology field. To understand how contact tracing influences a recovered phylogeny, we developed a new simulation tool called SEEPS (Sequence Evolution and Epidemiological Process Simulator) that allows for the simulation of contact tracing and the resulting transmission tree, pathogen phylogeny, and corresponding virus genetic sequences. Importantly, SEEPS takes within-host evolution into account when generating pathogen phylogenies and sequences from transmission histories. Using SEEPS, we demonstrate that contact tracing can significantly impact the structure of the resulting tree, as described by popular tree statistics. Contact tracing generates phylogenies that are less balanced than the underlying transmission process, less representative of the larger epidemiological process, and affects the internal/external branch length ratios that characterize specific epidemiological scenarios. We also examined real data from a 2007-2008 Swedish HIV-1 outbreak and the broader 1998-2010 European HIV-1 epidemic to highlight the differences in contact tracing and expected phylogenies. Aided by SEEPS, we show that the data collection of the Swedish outbreak was strongly influenced by contact tracing even after downsampling, while the broader European Union epidemic showed little evidence of universal contact tracing, agreeing with the known epidemiological information about sampling and spread. Overall, our results highlight the importance of including possible non-uniform sampling schemes when examining phylogenetic trees. For that, SEEPS serves as a useful tool to evaluate such impacts, thereby facilitating better phylogenetic inferences of the characteristics of a disease outbreak. SEEPS is available at https://github.com/MolEvolEpid/SEEPS.</p>\",\"PeriodicalId\":56026,\"journal\":{\"name\":\"Virus Evolution\",\"volume\":\"11 1\",\"pages\":\"veaf068\"},\"PeriodicalIF\":4.0000,\"publicationDate\":\"2025-09-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12481701/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Virus Evolution\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1093/ve/veaf068\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"VIROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Virus Evolution","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1093/ve/veaf068","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"VIROLOGY","Score":null,"Total":0}
Identifying impacts of contact tracing on HIV epidemiological inference from phylogenetic data.
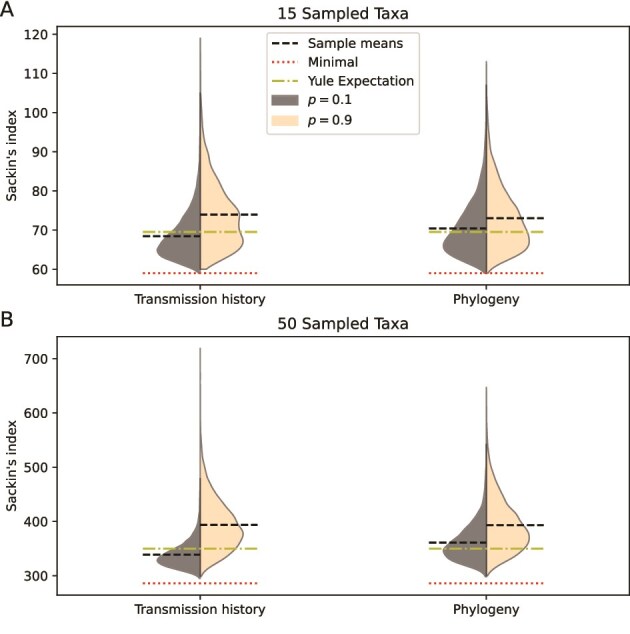
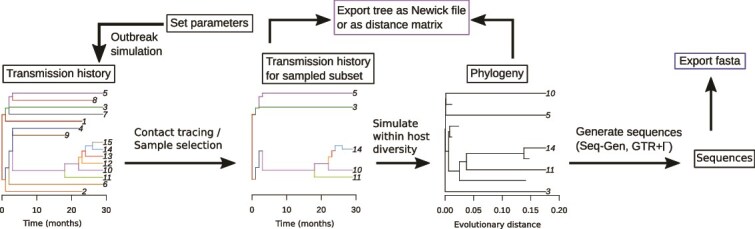
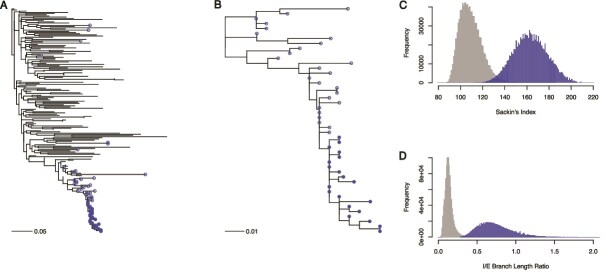
Robust sampling methods are foundational to inferences using phylogenies. Yet the impact of using contact tracing, a type of non-uniform sampling used in public health applications such as infectious disease outbreak investigations, has not been investigated in the molecular epidemiology field. To understand how contact tracing influences a recovered phylogeny, we developed a new simulation tool called SEEPS (Sequence Evolution and Epidemiological Process Simulator) that allows for the simulation of contact tracing and the resulting transmission tree, pathogen phylogeny, and corresponding virus genetic sequences. Importantly, SEEPS takes within-host evolution into account when generating pathogen phylogenies and sequences from transmission histories. Using SEEPS, we demonstrate that contact tracing can significantly impact the structure of the resulting tree, as described by popular tree statistics. Contact tracing generates phylogenies that are less balanced than the underlying transmission process, less representative of the larger epidemiological process, and affects the internal/external branch length ratios that characterize specific epidemiological scenarios. We also examined real data from a 2007-2008 Swedish HIV-1 outbreak and the broader 1998-2010 European HIV-1 epidemic to highlight the differences in contact tracing and expected phylogenies. Aided by SEEPS, we show that the data collection of the Swedish outbreak was strongly influenced by contact tracing even after downsampling, while the broader European Union epidemic showed little evidence of universal contact tracing, agreeing with the known epidemiological information about sampling and spread. Overall, our results highlight the importance of including possible non-uniform sampling schemes when examining phylogenetic trees. For that, SEEPS serves as a useful tool to evaluate such impacts, thereby facilitating better phylogenetic inferences of the characteristics of a disease outbreak. SEEPS is available at https://github.com/MolEvolEpid/SEEPS.
期刊介绍:
Virus Evolution is a new Open Access journal focusing on the long-term evolution of viruses, viruses as a model system for studying evolutionary processes, viral molecular epidemiology and environmental virology.
The aim of the journal is to provide a forum for original research papers, reviews, commentaries and a venue for in-depth discussion on the topics relevant to virus evolution.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


