Zhenzhu Zheng, Weilin Peng, Yiming Lin, Weihua Lin, Gaoxiong Wang
{"title":"中国4例全新羧化酶合成酶缺乏伴代谢性酸中毒的临床与遗传分析。","authors":"Zhenzhu Zheng, Weilin Peng, Yiming Lin, Weihua Lin, Gaoxiong Wang","doi":"10.1186/s13023-025-03723-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Holocarboxylase synthetase (HLCS) deficiency is an autosomal recessive organic acidaemia. This paper aimed to describe the clinical, biochemical and molecular features of four Chinese patients with HLCS deficiency, and to research the novel mutation.</p><p><strong>Methods: </strong>Tandem mass spectrometric analysis of elevated 3-hydroxyisovaleryl carnitine (C5OH) on dried blood spots was performed. Next-generation sequencing was then used to make a definite diagnosis, and the related variants were checked in several databases.</p><p><strong>Results: </strong>The four patients exhibited varying degrees of clinical symptoms, abnormal biochemical analysis and acylcarnitine profile. A total of six mutations in the HLCS gene were identified, including one novel missense mutation [c.1505 A > G (p.Gln502Arg)], and one frameshift mutation [c.2159delT (p.Leu720Profs*31)]. The variation c.1505 A > G (p.Gln502Arg) is predicted to be possibly damaging by several in silico prediction programs. Another frameshift variation, c.2159delT (p.Leu720Profs*31), is classified as uncertain significance.</p><p><strong>Conclusions: </strong>A novel variation c.1505 A > G (p.Gln502Arg) expands the mutational spectrum of the HLCS gene. Patient 4 is the first patient diagnosed as HLCS deficiency carrying the c.2159delT (p.Leu720Profs*31) variation. The results may contribute to a better understanding of the clinical course and genetic characteristics of patients with HLCS deficiency.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"491"},"PeriodicalIF":3.5000,"publicationDate":"2025-09-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12486680/pdf/","citationCount":"0","resultStr":"{\"title\":\"Clinical and genetic analysis of four Chinese patients with holocarboxylase synthetase deficiency and metabolic acidosis.\",\"authors\":\"Zhenzhu Zheng, Weilin Peng, Yiming Lin, Weihua Lin, Gaoxiong Wang\",\"doi\":\"10.1186/s13023-025-03723-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Holocarboxylase synthetase (HLCS) deficiency is an autosomal recessive organic acidaemia. This paper aimed to describe the clinical, biochemical and molecular features of four Chinese patients with HLCS deficiency, and to research the novel mutation.</p><p><strong>Methods: </strong>Tandem mass spectrometric analysis of elevated 3-hydroxyisovaleryl carnitine (C5OH) on dried blood spots was performed. Next-generation sequencing was then used to make a definite diagnosis, and the related variants were checked in several databases.</p><p><strong>Results: </strong>The four patients exhibited varying degrees of clinical symptoms, abnormal biochemical analysis and acylcarnitine profile. A total of six mutations in the HLCS gene were identified, including one novel missense mutation [c.1505 A > G (p.Gln502Arg)], and one frameshift mutation [c.2159delT (p.Leu720Profs*31)]. The variation c.1505 A > G (p.Gln502Arg) is predicted to be possibly damaging by several in silico prediction programs. Another frameshift variation, c.2159delT (p.Leu720Profs*31), is classified as uncertain significance.</p><p><strong>Conclusions: </strong>A novel variation c.1505 A > G (p.Gln502Arg) expands the mutational spectrum of the HLCS gene. Patient 4 is the first patient diagnosed as HLCS deficiency carrying the c.2159delT (p.Leu720Profs*31) variation. The results may contribute to a better understanding of the clinical course and genetic characteristics of patients with HLCS deficiency.</p>\",\"PeriodicalId\":19651,\"journal\":{\"name\":\"Orphanet Journal of Rare Diseases\",\"volume\":\"20 1\",\"pages\":\"491\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2025-09-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12486680/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Orphanet Journal of Rare Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13023-025-03723-2\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03723-2","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
背景:全新羧化酶合成酶缺乏症是一种常染色体隐性有机酸血症。本文旨在描述4例中国hcs缺乏症患者的临床、生化和分子特征,并对这种新的突变进行研究。方法:采用串联质谱法分析干血斑中3-羟基异戊基肉碱(C5OH)的含量。然后使用新一代测序做出明确的诊断,并在几个数据库中检查相关变异。结果:4例患者均表现出不同程度的临床症状,生化分析异常,酰基肉碱谱异常。共鉴定出6个HLCS基因突变,包括1个新的错义突变[c.1505]一个> G (p.Gln502Arg)]和一个移码突变[c]。2159年解决(p.Leu720Profs * 31)]。c.1505一个b> G (p.Gln502Arg)被几个计算机预测程序预测可能具有破坏性。另一个移码变异c.2159delT (p.Leu720Profs*31)被归为不确定显著性。结论:c.1505是一种新的变异b> G (p.Gln502Arg)扩展了HLCS基因的突变谱。患者4是第一个被诊断为携带c.2159delT (p.l u720profs *31)变异的HLCS缺陷患者。该结果可能有助于更好地了解HLCS缺乏症患者的临床病程和遗传特征。
Clinical and genetic analysis of four Chinese patients with holocarboxylase synthetase deficiency and metabolic acidosis.
Background: Holocarboxylase synthetase (HLCS) deficiency is an autosomal recessive organic acidaemia. This paper aimed to describe the clinical, biochemical and molecular features of four Chinese patients with HLCS deficiency, and to research the novel mutation.
Methods: Tandem mass spectrometric analysis of elevated 3-hydroxyisovaleryl carnitine (C5OH) on dried blood spots was performed. Next-generation sequencing was then used to make a definite diagnosis, and the related variants were checked in several databases.
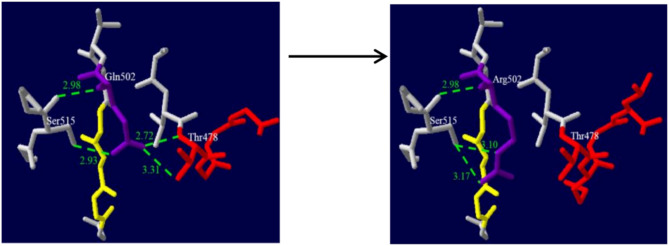
Results: The four patients exhibited varying degrees of clinical symptoms, abnormal biochemical analysis and acylcarnitine profile. A total of six mutations in the HLCS gene were identified, including one novel missense mutation [c.1505 A > G (p.Gln502Arg)], and one frameshift mutation [c.2159delT (p.Leu720Profs*31)]. The variation c.1505 A > G (p.Gln502Arg) is predicted to be possibly damaging by several in silico prediction programs. Another frameshift variation, c.2159delT (p.Leu720Profs*31), is classified as uncertain significance.
Conclusions: A novel variation c.1505 A > G (p.Gln502Arg) expands the mutational spectrum of the HLCS gene. Patient 4 is the first patient diagnosed as HLCS deficiency carrying the c.2159delT (p.Leu720Profs*31) variation. The results may contribute to a better understanding of the clinical course and genetic characteristics of patients with HLCS deficiency.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


