{"title":"常染色体显性多囊肾病和特发性精氨酸抗利尿素缺乏:一个加速肾功能衰退的特殊病例报告。","authors":"Farah Wehbe, Mark Elliott, Myriam Farah","doi":"10.1177/20543581251378016","DOIUrl":null,"url":null,"abstract":"<p><p>Autosomal dominant polycystic kidney disease (ADPKD) is a common genetic kidney disorder characterized by progressive cyst growth and kidney impairment. Arginine vasopressin deficiency (AVP-D) is a rare disorder resulting from reduced arginine vasopressin production, causing polyuria and thirst. The coexistence of ADPKD and AVP-D is rarely documented in the literature. We report what may be the first documented case of a patient diagnosed with ADPKD and idiopathic AVP-D. Initially managed with intranasal desmopressin, the patient's kidney function declined earlier than expected based on her ADPKD, progressing to kidney failure at a low total kidney volume (836 mL). This paradoxical outcome suggests that while AVP-D may have initially slowed cyst growth, her uncontrolled AVP-D likely contributed to kidney function decline, presumably due to recurrent volume depletion and acute kidney injuries. This case highlights the need for individualized AVP-D management in ADPKD patients and reiterates AVP's role in the complex pathophysiology of ADPKD progression.</p>","PeriodicalId":9426,"journal":{"name":"Canadian Journal of Kidney Health and Disease","volume":"12 ","pages":"20543581251378016"},"PeriodicalIF":1.5000,"publicationDate":"2025-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12477364/pdf/","citationCount":"0","resultStr":"{\"title\":\"Autosomal Dominant Polycystic Kidney Disease and Idiopathic Arginine Vasopressin Deficiency: A Peculiar Case Report of Accelerated Kidney Function Decline.\",\"authors\":\"Farah Wehbe, Mark Elliott, Myriam Farah\",\"doi\":\"10.1177/20543581251378016\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Autosomal dominant polycystic kidney disease (ADPKD) is a common genetic kidney disorder characterized by progressive cyst growth and kidney impairment. Arginine vasopressin deficiency (AVP-D) is a rare disorder resulting from reduced arginine vasopressin production, causing polyuria and thirst. The coexistence of ADPKD and AVP-D is rarely documented in the literature. We report what may be the first documented case of a patient diagnosed with ADPKD and idiopathic AVP-D. Initially managed with intranasal desmopressin, the patient's kidney function declined earlier than expected based on her ADPKD, progressing to kidney failure at a low total kidney volume (836 mL). This paradoxical outcome suggests that while AVP-D may have initially slowed cyst growth, her uncontrolled AVP-D likely contributed to kidney function decline, presumably due to recurrent volume depletion and acute kidney injuries. This case highlights the need for individualized AVP-D management in ADPKD patients and reiterates AVP's role in the complex pathophysiology of ADPKD progression.</p>\",\"PeriodicalId\":9426,\"journal\":{\"name\":\"Canadian Journal of Kidney Health and Disease\",\"volume\":\"12 \",\"pages\":\"20543581251378016\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2025-09-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12477364/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Canadian Journal of Kidney Health and Disease\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/20543581251378016\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"UROLOGY & NEPHROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Canadian Journal of Kidney Health and Disease","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/20543581251378016","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
Autosomal Dominant Polycystic Kidney Disease and Idiopathic Arginine Vasopressin Deficiency: A Peculiar Case Report of Accelerated Kidney Function Decline.
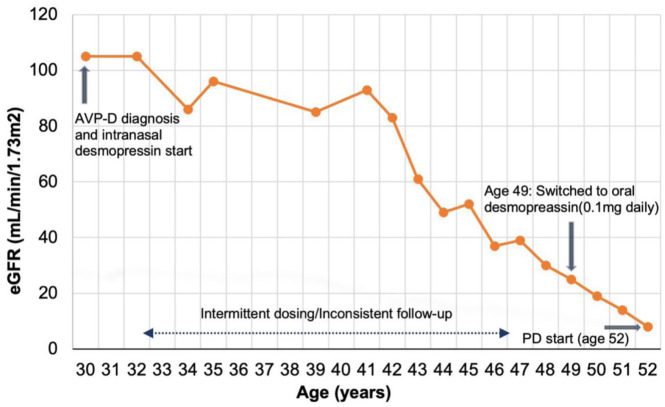
Autosomal dominant polycystic kidney disease (ADPKD) is a common genetic kidney disorder characterized by progressive cyst growth and kidney impairment. Arginine vasopressin deficiency (AVP-D) is a rare disorder resulting from reduced arginine vasopressin production, causing polyuria and thirst. The coexistence of ADPKD and AVP-D is rarely documented in the literature. We report what may be the first documented case of a patient diagnosed with ADPKD and idiopathic AVP-D. Initially managed with intranasal desmopressin, the patient's kidney function declined earlier than expected based on her ADPKD, progressing to kidney failure at a low total kidney volume (836 mL). This paradoxical outcome suggests that while AVP-D may have initially slowed cyst growth, her uncontrolled AVP-D likely contributed to kidney function decline, presumably due to recurrent volume depletion and acute kidney injuries. This case highlights the need for individualized AVP-D management in ADPKD patients and reiterates AVP's role in the complex pathophysiology of ADPKD progression.
期刊介绍:
Canadian Journal of Kidney Health and Disease, the official journal of the Canadian Society of Nephrology, is an open access, peer-reviewed online journal that encourages high quality submissions focused on clinical, translational and health services delivery research in the field of chronic kidney disease, dialysis, kidney transplantation and organ donation. Our mandate is to promote and advocate for kidney health as it impacts national and international communities. Basic science, translational studies and clinical studies will be peer reviewed and processed by an Editorial Board comprised of geographically diverse Canadian and international nephrologists, internists and allied health professionals; this Editorial Board is mandated to ensure highest quality publications.



 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


