Joanna Panecka-Hofman, Pasquale Linciano, Ina Pöhner, Edyta Dyguda-Kazimierowicz, Wiktoria Jedwabny, Giacomo Landi, Nuno Santarem, Gesa Witt, Bernhard Ellinger, Maria Kuzikov, Rosaria Luciani, Stefania Ferrari, Daniele Aiello, Stefano Mangani, Cecilia Pozzi, Anabela Cordeiro-da-Silva, Sheraz Gul, Maria Paola Costi, Rebecca C Wade
{"title":"多学科片段杂交方法设计靶向锥虫PTR1的2-氨基苯并噻唑衍生物。","authors":"Joanna Panecka-Hofman, Pasquale Linciano, Ina Pöhner, Edyta Dyguda-Kazimierowicz, Wiktoria Jedwabny, Giacomo Landi, Nuno Santarem, Gesa Witt, Bernhard Ellinger, Maria Kuzikov, Rosaria Luciani, Stefania Ferrari, Daniele Aiello, Stefano Mangani, Cecilia Pozzi, Anabela Cordeiro-da-Silva, Sheraz Gul, Maria Paola Costi, Rebecca C Wade","doi":"10.1021/acs.jmedchem.5c01799","DOIUrl":null,"url":null,"abstract":"<p><p>Pteridine reductase 1 (PTR1) is a folate pathway enzyme essential for pathogenic trypanosomatids and a promising drug target for diseases such as sleeping sickness and leishmaniasis. Previous studies have shown that the 2-aminobenzothiazole moiety targets the PTR1 biopterin pocket, while 3,4-dichlorophenyl-containing compounds, such as <b>I</b> bind a different region of the <i>Trypanosoma brucei</i> PTR1 (<i>Tb</i>PTR1) pocket. This study combines both moieties via various linkers, creating two compound series screened in silico against <i>Tb</i>PTR1 and <i>Leishmania major</i> PTR1 (<i>Lm</i>PTR1). In the first series, five compounds were synthesized, and <b>1a</b> and <b>1b</b> emerged as potent <i>Tb</i>PTR1 inhibitors, with <b>1b</b> also being active against <i>Lm</i>PTR1 and moderately effective against <i>Leishmania infantum</i>. Furthermore, structure-activity relationship analysis, supported by quantum calculations and crystallography, revealed meta-halogenation to be more favorable than para, although single halogenation reduced antiparasite effects. Our fragment hybridization approach led to less toxic, more effective compounds than <b>I</b>.</p>","PeriodicalId":46,"journal":{"name":"Journal of Medicinal Chemistry","volume":" ","pages":""},"PeriodicalIF":6.8000,"publicationDate":"2025-09-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Design of 2-Aminobenzothiazole Derivatives Targeting Trypanosomatid PTR1 by a Multidisciplinary Fragment Hybridization Approach.\",\"authors\":\"Joanna Panecka-Hofman, Pasquale Linciano, Ina Pöhner, Edyta Dyguda-Kazimierowicz, Wiktoria Jedwabny, Giacomo Landi, Nuno Santarem, Gesa Witt, Bernhard Ellinger, Maria Kuzikov, Rosaria Luciani, Stefania Ferrari, Daniele Aiello, Stefano Mangani, Cecilia Pozzi, Anabela Cordeiro-da-Silva, Sheraz Gul, Maria Paola Costi, Rebecca C Wade\",\"doi\":\"10.1021/acs.jmedchem.5c01799\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Pteridine reductase 1 (PTR1) is a folate pathway enzyme essential for pathogenic trypanosomatids and a promising drug target for diseases such as sleeping sickness and leishmaniasis. Previous studies have shown that the 2-aminobenzothiazole moiety targets the PTR1 biopterin pocket, while 3,4-dichlorophenyl-containing compounds, such as <b>I</b> bind a different region of the <i>Trypanosoma brucei</i> PTR1 (<i>Tb</i>PTR1) pocket. This study combines both moieties via various linkers, creating two compound series screened in silico against <i>Tb</i>PTR1 and <i>Leishmania major</i> PTR1 (<i>Lm</i>PTR1). In the first series, five compounds were synthesized, and <b>1a</b> and <b>1b</b> emerged as potent <i>Tb</i>PTR1 inhibitors, with <b>1b</b> also being active against <i>Lm</i>PTR1 and moderately effective against <i>Leishmania infantum</i>. Furthermore, structure-activity relationship analysis, supported by quantum calculations and crystallography, revealed meta-halogenation to be more favorable than para, although single halogenation reduced antiparasite effects. Our fragment hybridization approach led to less toxic, more effective compounds than <b>I</b>.</p>\",\"PeriodicalId\":46,\"journal\":{\"name\":\"Journal of Medicinal Chemistry\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":6.8000,\"publicationDate\":\"2025-09-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Medicinal Chemistry\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jmedchem.5c01799\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Medicinal Chemistry","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1021/acs.jmedchem.5c01799","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
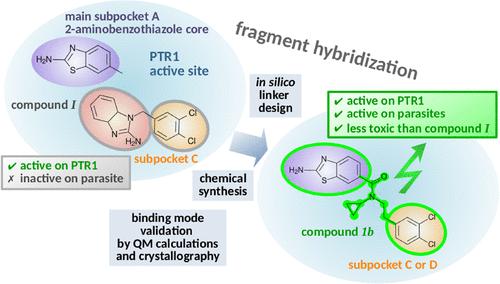
Design of 2-Aminobenzothiazole Derivatives Targeting Trypanosomatid PTR1 by a Multidisciplinary Fragment Hybridization Approach.
Pteridine reductase 1 (PTR1) is a folate pathway enzyme essential for pathogenic trypanosomatids and a promising drug target for diseases such as sleeping sickness and leishmaniasis. Previous studies have shown that the 2-aminobenzothiazole moiety targets the PTR1 biopterin pocket, while 3,4-dichlorophenyl-containing compounds, such as I bind a different region of the Trypanosoma brucei PTR1 (TbPTR1) pocket. This study combines both moieties via various linkers, creating two compound series screened in silico against TbPTR1 and Leishmania major PTR1 (LmPTR1). In the first series, five compounds were synthesized, and 1a and 1b emerged as potent TbPTR1 inhibitors, with 1b also being active against LmPTR1 and moderately effective against Leishmania infantum. Furthermore, structure-activity relationship analysis, supported by quantum calculations and crystallography, revealed meta-halogenation to be more favorable than para, although single halogenation reduced antiparasite effects. Our fragment hybridization approach led to less toxic, more effective compounds than I.
期刊介绍:
The Journal of Medicinal Chemistry is a prestigious biweekly peer-reviewed publication that focuses on the multifaceted field of medicinal chemistry. Since its inception in 1959 as the Journal of Medicinal and Pharmaceutical Chemistry, it has evolved to become a cornerstone in the dissemination of research findings related to the design, synthesis, and development of therapeutic agents.
The Journal of Medicinal Chemistry is recognized for its significant impact in the scientific community, as evidenced by its 2022 impact factor of 7.3. This metric reflects the journal's influence and the importance of its content in shaping the future of drug discovery and development. The journal serves as a vital resource for chemists, pharmacologists, and other researchers interested in the molecular mechanisms of drug action and the optimization of therapeutic compounds.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


