Lea M. Ibele, , , Carlo Adamo, , and , Davide Avagliano*,
{"title":"非绝热动力学中的基准密度泛函近似:视网膜模型中的反式顺式异构化。","authors":"Lea M. Ibele, , , Carlo Adamo, , and , Davide Avagliano*, ","doi":"10.1021/acs.jctc.5c01200","DOIUrl":null,"url":null,"abstract":"<p >An exhaustive benchmark of density functional approximations (DFAs) for nonadiabatic dynamics is reported on the <i>trans</i>–<i>cis</i> photoisomerization of the protonated Schiff base 3 (PSB3), which presents numerous challenges for time-dependent density functional theory (TD-DFT). We introduce a rigorous protocol for benchmarking DFAs for nonadiabatic dynamics regarding the initialization, the dynamics, and its evaluation. Different families of DFAs were compared with a high-level reference, highlighting that electronic populations are an unsuitable metric for evaluating the accuracy of dynamics. We found that several local functionals showed the best agreement of the population decay with the reference RMS-CASPT2, but strictly passed through a deactivation channel dominated by a single-bond torsion that is not accessible in the reference and in the literature. While using 100% Hartree–Fock exchange in the functional yields the only correct isomerization behavior, the time scales and quantum yields are far off the reference values, due to an artificial local minimum being predicted along the wrong torsion coordinate. Static energy scans suggest that this issue can be circumvented by double hybrid functionals, in particular those balancing nonlocal exchange and correlation combined with range-separation. Indeed, they predict energy profiles along the two torsion coordinates in close agreement with the RMS-CASPT2 reference. This emphasizes the impact these DFAs will have on the field of nonadiabatic dynamics once analytical gradients are introduced.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9799–9813"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Benchmarking Density Functional Approximations in Nonadiabatic Dynamics: Trans–Cis Isomerization in Retinal Model\",\"authors\":\"Lea M. Ibele, , , Carlo Adamo, , and , Davide Avagliano*, \",\"doi\":\"10.1021/acs.jctc.5c01200\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >An exhaustive benchmark of density functional approximations (DFAs) for nonadiabatic dynamics is reported on the <i>trans</i>–<i>cis</i> photoisomerization of the protonated Schiff base 3 (PSB3), which presents numerous challenges for time-dependent density functional theory (TD-DFT). We introduce a rigorous protocol for benchmarking DFAs for nonadiabatic dynamics regarding the initialization, the dynamics, and its evaluation. Different families of DFAs were compared with a high-level reference, highlighting that electronic populations are an unsuitable metric for evaluating the accuracy of dynamics. We found that several local functionals showed the best agreement of the population decay with the reference RMS-CASPT2, but strictly passed through a deactivation channel dominated by a single-bond torsion that is not accessible in the reference and in the literature. While using 100% Hartree–Fock exchange in the functional yields the only correct isomerization behavior, the time scales and quantum yields are far off the reference values, due to an artificial local minimum being predicted along the wrong torsion coordinate. Static energy scans suggest that this issue can be circumvented by double hybrid functionals, in particular those balancing nonlocal exchange and correlation combined with range-separation. Indeed, they predict energy profiles along the two torsion coordinates in close agreement with the RMS-CASPT2 reference. This emphasizes the impact these DFAs will have on the field of nonadiabatic dynamics once analytical gradients are introduced.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"21 19\",\"pages\":\"9799–9813\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-09-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01200\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01200","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
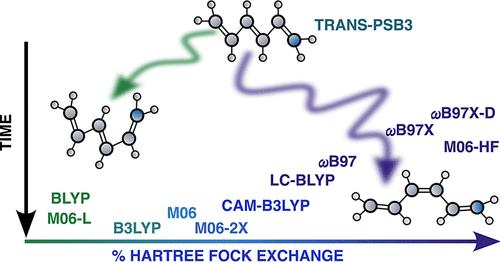
Benchmarking Density Functional Approximations in Nonadiabatic Dynamics: Trans–Cis Isomerization in Retinal Model
An exhaustive benchmark of density functional approximations (DFAs) for nonadiabatic dynamics is reported on the trans–cis photoisomerization of the protonated Schiff base 3 (PSB3), which presents numerous challenges for time-dependent density functional theory (TD-DFT). We introduce a rigorous protocol for benchmarking DFAs for nonadiabatic dynamics regarding the initialization, the dynamics, and its evaluation. Different families of DFAs were compared with a high-level reference, highlighting that electronic populations are an unsuitable metric for evaluating the accuracy of dynamics. We found that several local functionals showed the best agreement of the population decay with the reference RMS-CASPT2, but strictly passed through a deactivation channel dominated by a single-bond torsion that is not accessible in the reference and in the literature. While using 100% Hartree–Fock exchange in the functional yields the only correct isomerization behavior, the time scales and quantum yields are far off the reference values, due to an artificial local minimum being predicted along the wrong torsion coordinate. Static energy scans suggest that this issue can be circumvented by double hybrid functionals, in particular those balancing nonlocal exchange and correlation combined with range-separation. Indeed, they predict energy profiles along the two torsion coordinates in close agreement with the RMS-CASPT2 reference. This emphasizes the impact these DFAs will have on the field of nonadiabatic dynamics once analytical gradients are introduced.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


