{"title":"病例报告:MRPS36的双等位基因变异,编码2-氧戊二酸脱氢酶复合物的一个组成部分,导致leigh综合征。","authors":"Huafang Jiang, Chaolong Xu, Zhimei Liu, Ruoyu Duan, Xingfeng Yao, Xiaona Fu, Jiatong Xu, Xuejing Kang, Tenghui Yu, Yuanyuan Wang, Fang Fang","doi":"10.3389/fped.2025.1608840","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The <i>MRPS36</i> gene encodes the E4 subunit of the 2-oxoglutarate dehydrogenase complex (OGDHC), a critical enzyme in the tricarboxylic acid cycle. OGDHC deficiency can lead to metabolic disorders with a clinical spectrum ranging from fatal neonatal lactic acidosis to variable degrees of global developmental delay and movement disorders. To date, a homozygous <i>MRPS36</i> variant has been reported as a novel cause of Leigh syndrome in only two siblings, who presented with developmental delay, movement disorders, bilateral striatal necrosis, and reduced OGDHC activity.</p><p><strong>Case presentation: </strong>We report a third case of Leigh syndrome associated with <i>MRPS36</i> variants in a 2-year-old boy. The patient exhibited with global developmental delay, dystonia, early-onset chorea, and elevated serum lactate levels. Follow-up brain magnetic resonance imaging at 2 years revealed progressive degenerative lesions in the bilateral basal ganglia. Muscle biopsy showed abnormal mitochondrial accumulation beneath the sarcolemma, and the oxygen consumption rate was reduced in skin fibroblasts. Whole-exome sequencing identified two novel compound heterozygous <i>MRPS36</i> variants: c.42+1G>A (p.?) and c.296G>C (p.Arg99Pro).</p><p><strong>Conclusion: </strong>This case supports <i>MRPS36</i> as a novel pathogenic cause of Leigh syndrome, further expanding the genetic spectrum of the disorder. Key clinical features include developmental delay, involuntary movement disorders, progressive basal ganglia atrophy, and a slowly progressive disease course.</p>","PeriodicalId":12637,"journal":{"name":"Frontiers in Pediatrics","volume":"13 ","pages":"1608840"},"PeriodicalIF":2.0000,"publicationDate":"2025-09-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12463997/pdf/","citationCount":"0","resultStr":"{\"title\":\"Case Report: Biallelic variants in <i>MRPS36</i>, encoding a component of the 2-oxoglutarate dehydrogenase complex, cause leigh syndrome.\",\"authors\":\"Huafang Jiang, Chaolong Xu, Zhimei Liu, Ruoyu Duan, Xingfeng Yao, Xiaona Fu, Jiatong Xu, Xuejing Kang, Tenghui Yu, Yuanyuan Wang, Fang Fang\",\"doi\":\"10.3389/fped.2025.1608840\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The <i>MRPS36</i> gene encodes the E4 subunit of the 2-oxoglutarate dehydrogenase complex (OGDHC), a critical enzyme in the tricarboxylic acid cycle. OGDHC deficiency can lead to metabolic disorders with a clinical spectrum ranging from fatal neonatal lactic acidosis to variable degrees of global developmental delay and movement disorders. To date, a homozygous <i>MRPS36</i> variant has been reported as a novel cause of Leigh syndrome in only two siblings, who presented with developmental delay, movement disorders, bilateral striatal necrosis, and reduced OGDHC activity.</p><p><strong>Case presentation: </strong>We report a third case of Leigh syndrome associated with <i>MRPS36</i> variants in a 2-year-old boy. The patient exhibited with global developmental delay, dystonia, early-onset chorea, and elevated serum lactate levels. Follow-up brain magnetic resonance imaging at 2 years revealed progressive degenerative lesions in the bilateral basal ganglia. Muscle biopsy showed abnormal mitochondrial accumulation beneath the sarcolemma, and the oxygen consumption rate was reduced in skin fibroblasts. Whole-exome sequencing identified two novel compound heterozygous <i>MRPS36</i> variants: c.42+1G>A (p.?) and c.296G>C (p.Arg99Pro).</p><p><strong>Conclusion: </strong>This case supports <i>MRPS36</i> as a novel pathogenic cause of Leigh syndrome, further expanding the genetic spectrum of the disorder. Key clinical features include developmental delay, involuntary movement disorders, progressive basal ganglia atrophy, and a slowly progressive disease course.</p>\",\"PeriodicalId\":12637,\"journal\":{\"name\":\"Frontiers in Pediatrics\",\"volume\":\"13 \",\"pages\":\"1608840\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2025-09-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12463997/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Frontiers in Pediatrics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.3389/fped.2025.1608840\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"PEDIATRICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in Pediatrics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3389/fped.2025.1608840","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"PEDIATRICS","Score":null,"Total":0}
Case Report: Biallelic variants in MRPS36, encoding a component of the 2-oxoglutarate dehydrogenase complex, cause leigh syndrome.
Background: The MRPS36 gene encodes the E4 subunit of the 2-oxoglutarate dehydrogenase complex (OGDHC), a critical enzyme in the tricarboxylic acid cycle. OGDHC deficiency can lead to metabolic disorders with a clinical spectrum ranging from fatal neonatal lactic acidosis to variable degrees of global developmental delay and movement disorders. To date, a homozygous MRPS36 variant has been reported as a novel cause of Leigh syndrome in only two siblings, who presented with developmental delay, movement disorders, bilateral striatal necrosis, and reduced OGDHC activity.
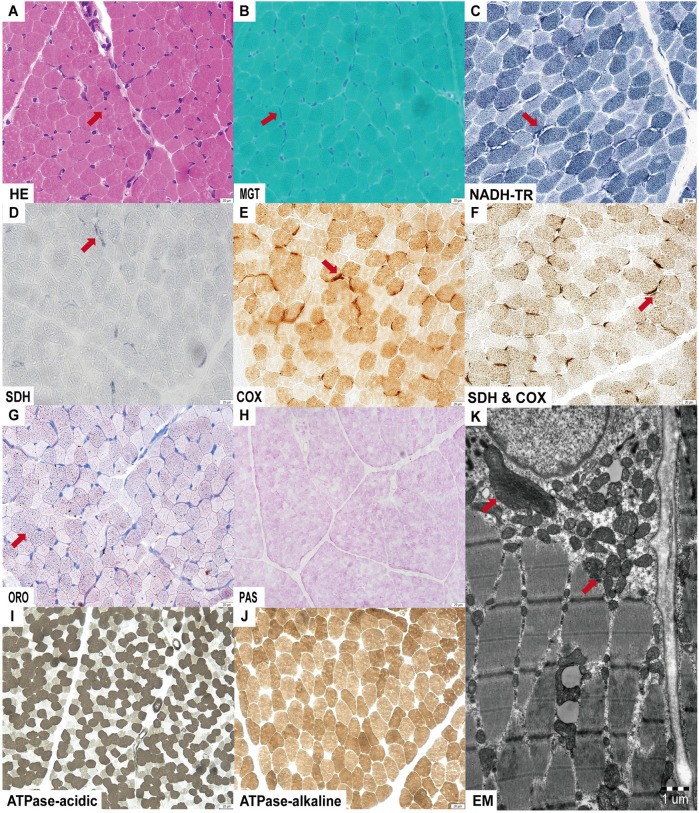
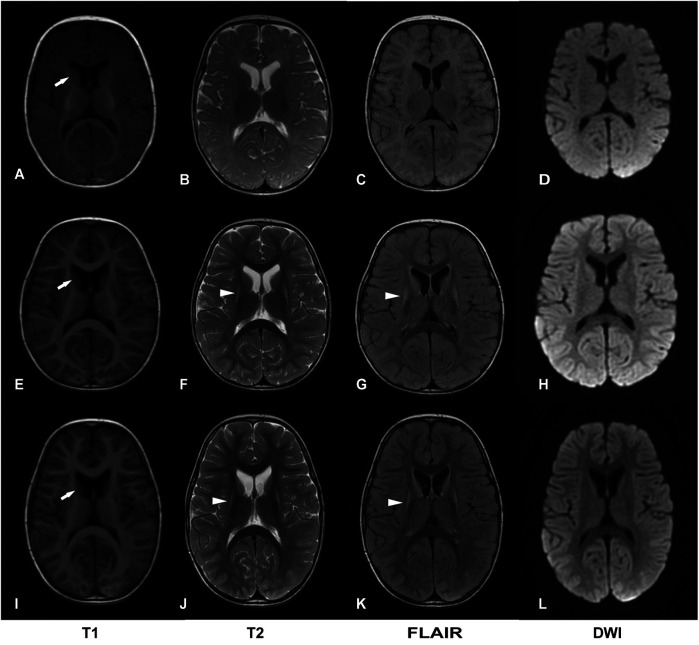
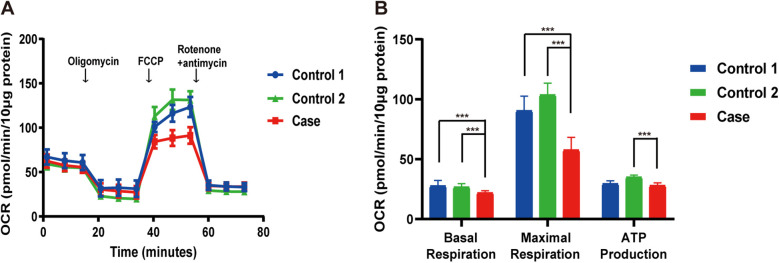
Case presentation: We report a third case of Leigh syndrome associated with MRPS36 variants in a 2-year-old boy. The patient exhibited with global developmental delay, dystonia, early-onset chorea, and elevated serum lactate levels. Follow-up brain magnetic resonance imaging at 2 years revealed progressive degenerative lesions in the bilateral basal ganglia. Muscle biopsy showed abnormal mitochondrial accumulation beneath the sarcolemma, and the oxygen consumption rate was reduced in skin fibroblasts. Whole-exome sequencing identified two novel compound heterozygous MRPS36 variants: c.42+1G>A (p.?) and c.296G>C (p.Arg99Pro).
Conclusion: This case supports MRPS36 as a novel pathogenic cause of Leigh syndrome, further expanding the genetic spectrum of the disorder. Key clinical features include developmental delay, involuntary movement disorders, progressive basal ganglia atrophy, and a slowly progressive disease course.
期刊介绍:
Frontiers in Pediatrics (Impact Factor 2.33) publishes rigorously peer-reviewed research broadly across the field, from basic to clinical research that meets ongoing challenges in pediatric patient care and child health. Field Chief Editors Arjan Te Pas at Leiden University and Michael L. Moritz at the Children''s Hospital of Pittsburgh are supported by an outstanding Editorial Board of international experts. This multidisciplinary open-access journal is at the forefront of disseminating and communicating scientific knowledge and impactful discoveries to researchers, academics, clinicians and the public worldwide.
Frontiers in Pediatrics also features Research Topics, Frontiers special theme-focused issues managed by Guest Associate Editors, addressing important areas in pediatrics. In this fashion, Frontiers serves as an outlet to publish the broadest aspects of pediatrics in both basic and clinical research, including high-quality reviews, case reports, editorials and commentaries related to all aspects of pediatrics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


