József Csóka*, , , Dénes Berta, , and , Péter R. Nagy*,
{"title":"基于局部量子嵌入的GGA代价下的混合DFT质量热化学和环境效应。","authors":"József Csóka*, , , Dénes Berta, , and , Péter R. Nagy*, ","doi":"10.1021/acs.jctc.5c01121","DOIUrl":null,"url":null,"abstract":"<p >Reliable thermochemical modeling of reaction mechanisms requires hybrid DFT or higher-level models as well as inclusion of environment, conformer, thermal, etc. effects. Quantum embedding, such as the Huzinaga-equation and projection-based models employed here, can make such computations more accessible by focusing the use of the more costly models to the atoms involved in forming and breaking the bonds or residing in interacting surfaces, etc. Here, we further accelerate these embedding computations by combining local approximations in the atomic orbital and auxiliary function space of the hybrid DFT part with a new in-core density fitting implementation optimized for multilayer DFT. The so introduced local embedded subsystem (LESS) framework, when increasing the size of the environment, leads to asymptotically constant cost for the hybrid DFT layer. We demonstrate on reaction and activation energies of practical homogeneous, heterogeneous and enzymatic catalysis reactions that the intrinsic accuracy of hybrid DFT is retained, with a few tenths of a kcal/mol error including all (embedding and local) approximations. Compared to the same complete (density fitted) hybrid DFT reference, the LESS hybrid DFT-in-GGA runtimes are 30–90 times faster on systems with up to 171–238 atoms. Achieving energetics with practically hybrid DFT quality and GGA cost is a significant step toward predictive thermochemistry including reliable sampling, dynamics, etc. as well as quantum environment effects.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9573–9586"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/pdf/10.1021/acs.jctc.5c01121","citationCount":"0","resultStr":"{\"title\":\"Hybrid DFT Quality Thermochemistry and Environment Effects at GGA Cost via Local Quantum Embedding\",\"authors\":\"József Csóka*, , , Dénes Berta, , and , Péter R. Nagy*, \",\"doi\":\"10.1021/acs.jctc.5c01121\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Reliable thermochemical modeling of reaction mechanisms requires hybrid DFT or higher-level models as well as inclusion of environment, conformer, thermal, etc. effects. Quantum embedding, such as the Huzinaga-equation and projection-based models employed here, can make such computations more accessible by focusing the use of the more costly models to the atoms involved in forming and breaking the bonds or residing in interacting surfaces, etc. Here, we further accelerate these embedding computations by combining local approximations in the atomic orbital and auxiliary function space of the hybrid DFT part with a new in-core density fitting implementation optimized for multilayer DFT. The so introduced local embedded subsystem (LESS) framework, when increasing the size of the environment, leads to asymptotically constant cost for the hybrid DFT layer. We demonstrate on reaction and activation energies of practical homogeneous, heterogeneous and enzymatic catalysis reactions that the intrinsic accuracy of hybrid DFT is retained, with a few tenths of a kcal/mol error including all (embedding and local) approximations. Compared to the same complete (density fitted) hybrid DFT reference, the LESS hybrid DFT-in-GGA runtimes are 30–90 times faster on systems with up to 171–238 atoms. Achieving energetics with practically hybrid DFT quality and GGA cost is a significant step toward predictive thermochemistry including reliable sampling, dynamics, etc. as well as quantum environment effects.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"21 19\",\"pages\":\"9573–9586\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-09-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/pdf/10.1021/acs.jctc.5c01121\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01121\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01121","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Hybrid DFT Quality Thermochemistry and Environment Effects at GGA Cost via Local Quantum Embedding
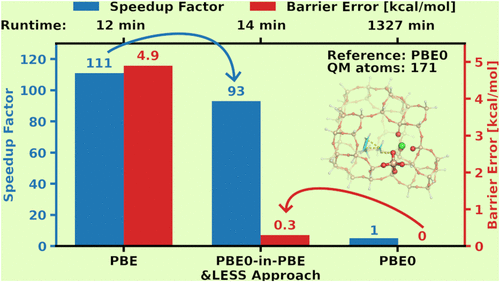
Reliable thermochemical modeling of reaction mechanisms requires hybrid DFT or higher-level models as well as inclusion of environment, conformer, thermal, etc. effects. Quantum embedding, such as the Huzinaga-equation and projection-based models employed here, can make such computations more accessible by focusing the use of the more costly models to the atoms involved in forming and breaking the bonds or residing in interacting surfaces, etc. Here, we further accelerate these embedding computations by combining local approximations in the atomic orbital and auxiliary function space of the hybrid DFT part with a new in-core density fitting implementation optimized for multilayer DFT. The so introduced local embedded subsystem (LESS) framework, when increasing the size of the environment, leads to asymptotically constant cost for the hybrid DFT layer. We demonstrate on reaction and activation energies of practical homogeneous, heterogeneous and enzymatic catalysis reactions that the intrinsic accuracy of hybrid DFT is retained, with a few tenths of a kcal/mol error including all (embedding and local) approximations. Compared to the same complete (density fitted) hybrid DFT reference, the LESS hybrid DFT-in-GGA runtimes are 30–90 times faster on systems with up to 171–238 atoms. Achieving energetics with practically hybrid DFT quality and GGA cost is a significant step toward predictive thermochemistry including reliable sampling, dynamics, etc. as well as quantum environment effects.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


