Nikhil Yenugu, , , Ashwani K. Tiwari*, , and , Sangita Sen*,
{"title":"强磁场中双原子分子Schrödinger方程的网格尺度不变非微扰解。","authors":"Nikhil Yenugu, , , Ashwani K. Tiwari*, , and , Sangita Sen*, ","doi":"10.1021/acs.jctc.5c00972","DOIUrl":null,"url":null,"abstract":"<p >The gauge invariant Wilson Hamiltonian is employed to solve the nuclear Schrödinger equation in the presence of a strong time-independent magnetic field. The single particle Hamiltonian is adapted to two particles in an external potential with the goal of computing the rovibrational spectra of diatomic molecules under various magnetic fields. A formalism involving reduced mass along with mass-weighted charges is presented. The Hamiltonian has its roots in Wilson’s lattice gauge theory, and we adopt a diagonalization-based nonperturbative algorithm with no limits on the range of applicability with respect to the strength of the magnetic field. We validate and benchmark our implementation by applying it to the extensively studied 2D single electron GaAs quantum dot subject to perpendicular uniform magnetic fields of strengths ranging from weak to ultrastrong (Zeeman–Landau regime), for which the analytical solutions are known. Our reduced-mass-reduced-charge formalism is then applied to compute the first few rovibrational states of a H<sub>2</sub> molecule modeled as a 2D harmonic oscillator subject to a perpendicular magnetic field and benchmarked against the corresponding analytical model. Our numerical method, it may be emphasized, can work with any binding potential supplied on a grid such as a Born–Oppenheimer potential energy surface with the field applied in any direction. To the best of our knowledge, this is the first application of the Wilson Hamiltonian to the computation of rovibrational spectra of molecules in magnetic fields and will allow a fully quantum and gauge invariant computation of diatomic rovibrational spectra in magnetic fields of arbitrary strengths and orientations.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9753–9771"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A Grid-Based Gauge-Invariant Non-Perturbative Solution of the Schrödinger Equation for Diatomic Molecules in Strong Magnetic Fields\",\"authors\":\"Nikhil Yenugu, , , Ashwani K. Tiwari*, , and , Sangita Sen*, \",\"doi\":\"10.1021/acs.jctc.5c00972\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The gauge invariant Wilson Hamiltonian is employed to solve the nuclear Schrödinger equation in the presence of a strong time-independent magnetic field. The single particle Hamiltonian is adapted to two particles in an external potential with the goal of computing the rovibrational spectra of diatomic molecules under various magnetic fields. A formalism involving reduced mass along with mass-weighted charges is presented. The Hamiltonian has its roots in Wilson’s lattice gauge theory, and we adopt a diagonalization-based nonperturbative algorithm with no limits on the range of applicability with respect to the strength of the magnetic field. We validate and benchmark our implementation by applying it to the extensively studied 2D single electron GaAs quantum dot subject to perpendicular uniform magnetic fields of strengths ranging from weak to ultrastrong (Zeeman–Landau regime), for which the analytical solutions are known. Our reduced-mass-reduced-charge formalism is then applied to compute the first few rovibrational states of a H<sub>2</sub> molecule modeled as a 2D harmonic oscillator subject to a perpendicular magnetic field and benchmarked against the corresponding analytical model. Our numerical method, it may be emphasized, can work with any binding potential supplied on a grid such as a Born–Oppenheimer potential energy surface with the field applied in any direction. To the best of our knowledge, this is the first application of the Wilson Hamiltonian to the computation of rovibrational spectra of molecules in magnetic fields and will allow a fully quantum and gauge invariant computation of diatomic rovibrational spectra in magnetic fields of arbitrary strengths and orientations.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"21 19\",\"pages\":\"9753–9771\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-09-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00972\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00972","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
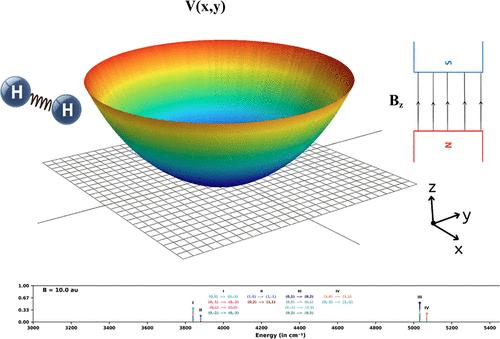
A Grid-Based Gauge-Invariant Non-Perturbative Solution of the Schrödinger Equation for Diatomic Molecules in Strong Magnetic Fields
The gauge invariant Wilson Hamiltonian is employed to solve the nuclear Schrödinger equation in the presence of a strong time-independent magnetic field. The single particle Hamiltonian is adapted to two particles in an external potential with the goal of computing the rovibrational spectra of diatomic molecules under various magnetic fields. A formalism involving reduced mass along with mass-weighted charges is presented. The Hamiltonian has its roots in Wilson’s lattice gauge theory, and we adopt a diagonalization-based nonperturbative algorithm with no limits on the range of applicability with respect to the strength of the magnetic field. We validate and benchmark our implementation by applying it to the extensively studied 2D single electron GaAs quantum dot subject to perpendicular uniform magnetic fields of strengths ranging from weak to ultrastrong (Zeeman–Landau regime), for which the analytical solutions are known. Our reduced-mass-reduced-charge formalism is then applied to compute the first few rovibrational states of a H2 molecule modeled as a 2D harmonic oscillator subject to a perpendicular magnetic field and benchmarked against the corresponding analytical model. Our numerical method, it may be emphasized, can work with any binding potential supplied on a grid such as a Born–Oppenheimer potential energy surface with the field applied in any direction. To the best of our knowledge, this is the first application of the Wilson Hamiltonian to the computation of rovibrational spectra of molecules in magnetic fields and will allow a fully quantum and gauge invariant computation of diatomic rovibrational spectra in magnetic fields of arbitrary strengths and orientations.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


