Roman Bykov, Tarek Itani, Daria Pletenchuk, Olesia Ohlopkova, Alexey Moshkin, Marina Stepanyuk, Aleksandr Semenov
{"title":"俄罗斯联邦斯维尔德洛夫斯克地区人类诺如病毒的分子特征和流行病学。","authors":"Roman Bykov, Tarek Itani, Daria Pletenchuk, Olesia Ohlopkova, Alexey Moshkin, Marina Stepanyuk, Aleksandr Semenov","doi":"10.3390/v17091243","DOIUrl":null,"url":null,"abstract":"<p><p>Human noroviruses (HuNoVs) stand as the primary cause of acute viral gastroenteritis outbreaks worldwide, particularly impacting children under the age of five. In Russia, reports of norovirus gastroenteritis have surged, especially in the post-COVID-19 era starting in 2022, with elevated infection rates reported into 2024. These viruses exhibit significant mutational variability, leading to the emergence of recombinant strains that can evade immune responses. A comprehensive examination of the complete genome is crucial for understanding the evolution of norovirus genes and for predicting potential outbreaks. This research focuses on analyzing the genotypic composition of HuNoVs circulating in the Sverdlovsk region during 2024, using Sanger sequencing and next-generation sequencing (NGS). Biological samples were collected (n = 384) from patients diagnosed with norovirus infection within the region. Bioinformatics analysis targeted the nucleotide sequences of the ORF1/ORF2 fragment and the assembly of complete genomes for the GII.4 and GII.7 genotypes. In total, 220 HuNoVs were characterized, representing 57.3% of the collected samples. The main capsid variants forming the predominant genotypic profile included GII.4 (n = 88, 40%), GII.7 (n = 86, 39%), and GII.17 (n = 14, 6%). Using NGS, we successfully assembled 8 out of 10 complete genomes for noroviruses GII.4[P16] and GII.7[P7]. Non-synonymous substitutions appeared at amino acid sites corresponding to the subdomains of VP1 in these strains. This molecular-genetic analysis provides contemporary insights into the genotypic composition, circulation patterns, and evolutionary dynamics associated with the dominant genovariants GII.4[P16] and GII.7[P7].</p>","PeriodicalId":49328,"journal":{"name":"Viruses-Basel","volume":"17 9","pages":""},"PeriodicalIF":3.5000,"publicationDate":"2025-09-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12474444/pdf/","citationCount":"0","resultStr":"{\"title\":\"Molecular Characterization and Epidemiology of Human Noroviruses in the Sverdlovsk Region, Russian Federation.\",\"authors\":\"Roman Bykov, Tarek Itani, Daria Pletenchuk, Olesia Ohlopkova, Alexey Moshkin, Marina Stepanyuk, Aleksandr Semenov\",\"doi\":\"10.3390/v17091243\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Human noroviruses (HuNoVs) stand as the primary cause of acute viral gastroenteritis outbreaks worldwide, particularly impacting children under the age of five. In Russia, reports of norovirus gastroenteritis have surged, especially in the post-COVID-19 era starting in 2022, with elevated infection rates reported into 2024. These viruses exhibit significant mutational variability, leading to the emergence of recombinant strains that can evade immune responses. A comprehensive examination of the complete genome is crucial for understanding the evolution of norovirus genes and for predicting potential outbreaks. This research focuses on analyzing the genotypic composition of HuNoVs circulating in the Sverdlovsk region during 2024, using Sanger sequencing and next-generation sequencing (NGS). Biological samples were collected (n = 384) from patients diagnosed with norovirus infection within the region. Bioinformatics analysis targeted the nucleotide sequences of the ORF1/ORF2 fragment and the assembly of complete genomes for the GII.4 and GII.7 genotypes. In total, 220 HuNoVs were characterized, representing 57.3% of the collected samples. The main capsid variants forming the predominant genotypic profile included GII.4 (n = 88, 40%), GII.7 (n = 86, 39%), and GII.17 (n = 14, 6%). Using NGS, we successfully assembled 8 out of 10 complete genomes for noroviruses GII.4[P16] and GII.7[P7]. Non-synonymous substitutions appeared at amino acid sites corresponding to the subdomains of VP1 in these strains. This molecular-genetic analysis provides contemporary insights into the genotypic composition, circulation patterns, and evolutionary dynamics associated with the dominant genovariants GII.4[P16] and GII.7[P7].</p>\",\"PeriodicalId\":49328,\"journal\":{\"name\":\"Viruses-Basel\",\"volume\":\"17 9\",\"pages\":\"\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2025-09-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12474444/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Viruses-Basel\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.3390/v17091243\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"VIROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Viruses-Basel","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3390/v17091243","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"VIROLOGY","Score":null,"Total":0}
Molecular Characterization and Epidemiology of Human Noroviruses in the Sverdlovsk Region, Russian Federation.
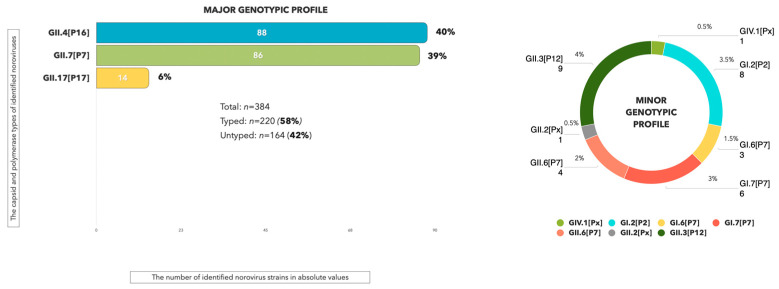
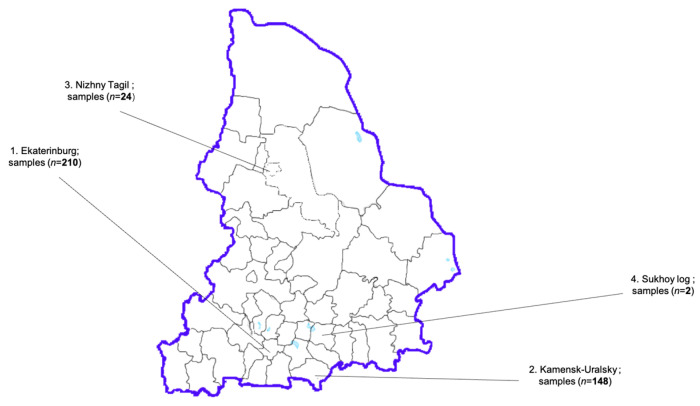
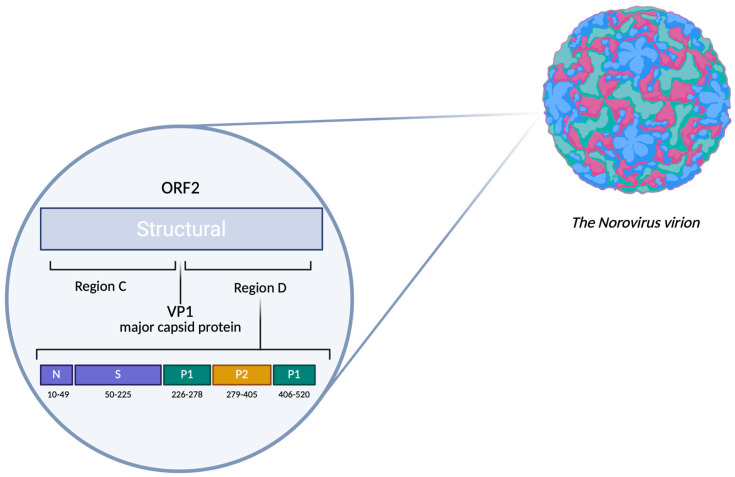
Human noroviruses (HuNoVs) stand as the primary cause of acute viral gastroenteritis outbreaks worldwide, particularly impacting children under the age of five. In Russia, reports of norovirus gastroenteritis have surged, especially in the post-COVID-19 era starting in 2022, with elevated infection rates reported into 2024. These viruses exhibit significant mutational variability, leading to the emergence of recombinant strains that can evade immune responses. A comprehensive examination of the complete genome is crucial for understanding the evolution of norovirus genes and for predicting potential outbreaks. This research focuses on analyzing the genotypic composition of HuNoVs circulating in the Sverdlovsk region during 2024, using Sanger sequencing and next-generation sequencing (NGS). Biological samples were collected (n = 384) from patients diagnosed with norovirus infection within the region. Bioinformatics analysis targeted the nucleotide sequences of the ORF1/ORF2 fragment and the assembly of complete genomes for the GII.4 and GII.7 genotypes. In total, 220 HuNoVs were characterized, representing 57.3% of the collected samples. The main capsid variants forming the predominant genotypic profile included GII.4 (n = 88, 40%), GII.7 (n = 86, 39%), and GII.17 (n = 14, 6%). Using NGS, we successfully assembled 8 out of 10 complete genomes for noroviruses GII.4[P16] and GII.7[P7]. Non-synonymous substitutions appeared at amino acid sites corresponding to the subdomains of VP1 in these strains. This molecular-genetic analysis provides contemporary insights into the genotypic composition, circulation patterns, and evolutionary dynamics associated with the dominant genovariants GII.4[P16] and GII.7[P7].
期刊介绍:
Viruses (ISSN 1999-4915) is an open access journal which provides an advanced forum for studies of viruses. It publishes reviews, regular research papers, communications, conference reports and short notes. Our aim is to encourage scientists to publish their experimental and theoretical results in as much detail as possible. There is no restriction on the length of the papers. The full experimental details must be provided so that the results can be reproduced. We also encourage the publication of timely reviews and commentaries on topics of interest to the virology community and feature highlights from the virology literature in the ''News and Views'' section. Electronic files or software regarding the full details of the calculation and experimental procedure, if unable to be published in a normal way, can be deposited as supplementary material.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


