Ali Albaghdadi, Ali Bakhshayeshi, Rohollah Taghavimendi, Leili Motevalizadeh
{"title":"利用密度泛函理论方法计算K3GaF6结构的结构、电子和光学性质的第一性原理","authors":"Ali Albaghdadi, Ali Bakhshayeshi, Rohollah Taghavimendi, Leili Motevalizadeh","doi":"10.1140/epjb/s10051-025-01014-0","DOIUrl":null,"url":null,"abstract":"<div><p>The structural, electronic, and optical properties of the K<sub>3</sub>GaF<sub>6</sub> compound were investigated using the Wien2k package, based on the density functional theory (DFT) and the full-potential linearized augmented plane-wave (FP-LAPW) method. The atomic positions in the structure were optimized, and the generalized gradient approximation (GGA) was employed for the exchange–correlation functional. The total and partial densities of states (DOS), Van Hove singularities, and the electronic contributions from each atom within specific energy ranges were analyzed in detail. The calculated band structure along the high-symmetry directions in the Brillouin zone reveals a direct bandgap of approximately 5.58 eV, primarily arising from the hybridization of atomic orbitals in the conduction band. In the optical analysis, the static dielectric constant, as well as the real and imaginary parts of the dielectric function, and the reflectivity were computed. K<sub>3</sub>GaF<sub>6</sub> exhibits low reflectivity and high transparency across a wide range of photon energies, making it a promising candidate for optoelectronic and UV-transparent applications.</p><h3>Graphical abstract</h3><div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>","PeriodicalId":787,"journal":{"name":"The European Physical Journal B","volume":"98 8","pages":""},"PeriodicalIF":1.7000,"publicationDate":"2025-08-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"First principles calculations of structural, electronic, and optical properties of K3GaF6 structure using density functional theory approach\",\"authors\":\"Ali Albaghdadi, Ali Bakhshayeshi, Rohollah Taghavimendi, Leili Motevalizadeh\",\"doi\":\"10.1140/epjb/s10051-025-01014-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>The structural, electronic, and optical properties of the K<sub>3</sub>GaF<sub>6</sub> compound were investigated using the Wien2k package, based on the density functional theory (DFT) and the full-potential linearized augmented plane-wave (FP-LAPW) method. The atomic positions in the structure were optimized, and the generalized gradient approximation (GGA) was employed for the exchange–correlation functional. The total and partial densities of states (DOS), Van Hove singularities, and the electronic contributions from each atom within specific energy ranges were analyzed in detail. The calculated band structure along the high-symmetry directions in the Brillouin zone reveals a direct bandgap of approximately 5.58 eV, primarily arising from the hybridization of atomic orbitals in the conduction band. In the optical analysis, the static dielectric constant, as well as the real and imaginary parts of the dielectric function, and the reflectivity were computed. K<sub>3</sub>GaF<sub>6</sub> exhibits low reflectivity and high transparency across a wide range of photon energies, making it a promising candidate for optoelectronic and UV-transparent applications.</p><h3>Graphical abstract</h3><div><figure><div><div><picture><source><img></source></picture></div></div></figure></div></div>\",\"PeriodicalId\":787,\"journal\":{\"name\":\"The European Physical Journal B\",\"volume\":\"98 8\",\"pages\":\"\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2025-08-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The European Physical Journal B\",\"FirstCategoryId\":\"4\",\"ListUrlMain\":\"https://link.springer.com/article/10.1140/epjb/s10051-025-01014-0\",\"RegionNum\":4,\"RegionCategory\":\"物理与天体物理\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"PHYSICS, CONDENSED MATTER\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The European Physical Journal B","FirstCategoryId":"4","ListUrlMain":"https://link.springer.com/article/10.1140/epjb/s10051-025-01014-0","RegionNum":4,"RegionCategory":"物理与天体物理","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PHYSICS, CONDENSED MATTER","Score":null,"Total":0}
First principles calculations of structural, electronic, and optical properties of K3GaF6 structure using density functional theory approach
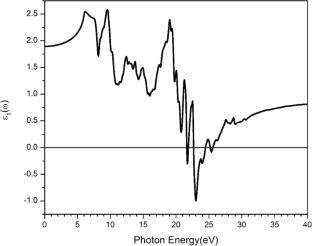
The structural, electronic, and optical properties of the K3GaF6 compound were investigated using the Wien2k package, based on the density functional theory (DFT) and the full-potential linearized augmented plane-wave (FP-LAPW) method. The atomic positions in the structure were optimized, and the generalized gradient approximation (GGA) was employed for the exchange–correlation functional. The total and partial densities of states (DOS), Van Hove singularities, and the electronic contributions from each atom within specific energy ranges were analyzed in detail. The calculated band structure along the high-symmetry directions in the Brillouin zone reveals a direct bandgap of approximately 5.58 eV, primarily arising from the hybridization of atomic orbitals in the conduction band. In the optical analysis, the static dielectric constant, as well as the real and imaginary parts of the dielectric function, and the reflectivity were computed. K3GaF6 exhibits low reflectivity and high transparency across a wide range of photon energies, making it a promising candidate for optoelectronic and UV-transparent applications.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


