氢键网络中氟诱导的扰动:来自FT-IR,拉曼和AIMD模拟的见解
IF 4.6
2区 化学
Q1 SPECTROSCOPY
Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy
Pub Date : 2025-09-24
DOI:10.1016/j.saa.2025.126994
引用次数: 0
摘要
氢键(HBs)控制着分子识别、催化和自组装,但它们的强度可以通过电子密度分布的细微变化而显著调节。氟,由于其高电负性和低极化率,以一种独特的位置依赖方式扰动HB网络。我们提出了一种集成的振动光谱(IR和拉曼)和从头算分子动力学(AIMD)方案,用于量化溶液中HB供体和受体官能团的强度。在HB供体溶剂(CH₃OH)和受体溶剂(DMSO)中研究了单氟苯胺异构体。Meta -氟对电子密度的吸收最强烈,使O-D···N相互作用减弱Δν = +22 cm−1,同时使DMSO中的NH给能增强Δν = 3 cm−1。邻位氟增加了受体强度(- 1 cm−1 in - od),但其分子内N-H··F键减少了分子间N-H··O键。对氟使两种模式基本不变。该工作流提供溶剂解析的HB参数,可以直接输入药效团评分,下一代力场参数化,以及任何需要准确供体/受体描述符的建模框架-从药物-靶标对接,通过电解质和聚合物设计,到超分子主-客工程和催化反应建模。本文章由计算机程序翻译,如有差异,请以英文原文为准。
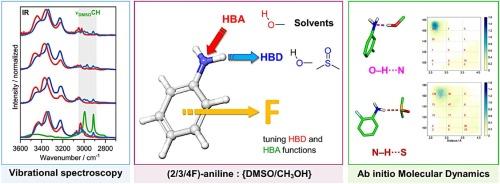
Fluorine-induced perturbations in hydrogen bond networks: Insights from FT-IR, Raman, and AIMD simulations
Hydrogen bonds (HBs) govern molecular recognition, catalysis, and self−assembly, yet their strength can be tuned dramatically by subtle changes in electron density distribution. Fluorine, owing to its high electronegativity and low polarizability, perturbs HB networks in a uniquely position−dependent manner. We present an integrated vibrational spectroscopy (IR and Raman) and ab initio molecular dynamics (AIMD) protocol that quantifies HB donor and acceptor strengths of functional groups in solution. Monofluoroaniline isomers were investigated in an HB donor solvent (CH₃OH) and an acceptor solvent (DMSO). Meta − fluorine withdraws electron density most strongly, weakening O–D···N interactions by Δν = +22 cm−1 and simultaneously enhancing N![]() H donation in DMSO by Δν = 3 cm−1. Ortho − fluorine increases acceptor strength (−1 cm−1 in νOD) but its intramolecular N–H···F contact reduces intermolecular N–H···O bonding. Para − fluorine leaves both modes essentially unchanged. The workflow furnishes solvent−resolved HB parameters that can feed directly into pharmacophore scoring, next−generation force−field parametrization, and any modeling framework that requires accurate donor/acceptor descriptors – ranging from drug−target docking, through electrolyte and polymer design, to supramolecular host−guest engineering and catalytic reaction modeling.
H donation in DMSO by Δν = 3 cm−1. Ortho − fluorine increases acceptor strength (−1 cm−1 in νOD) but its intramolecular N–H···F contact reduces intermolecular N–H···O bonding. Para − fluorine leaves both modes essentially unchanged. The workflow furnishes solvent−resolved HB parameters that can feed directly into pharmacophore scoring, next−generation force−field parametrization, and any modeling framework that requires accurate donor/acceptor descriptors – ranging from drug−target docking, through electrolyte and polymer design, to supramolecular host−guest engineering and catalytic reaction modeling.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
8.40
自引率
11.40%
发文量
1364
审稿时长
40 days
期刊介绍:
Spectrochimica Acta, Part A: Molecular and Biomolecular Spectroscopy (SAA) is an interdisciplinary journal which spans from basic to applied aspects of optical spectroscopy in chemistry, medicine, biology, and materials science.
The journal publishes original scientific papers that feature high-quality spectroscopic data and analysis. From the broad range of optical spectroscopies, the emphasis is on electronic, vibrational or rotational spectra of molecules, rather than on spectroscopy based on magnetic moments.
Criteria for publication in SAA are novelty, uniqueness, and outstanding quality. Routine applications of spectroscopic techniques and computational methods are not appropriate.
Topics of particular interest of Spectrochimica Acta Part A include, but are not limited to:
Spectroscopy and dynamics of bioanalytical, biomedical, environmental, and atmospheric sciences,
Novel experimental techniques or instrumentation for molecular spectroscopy,
Novel theoretical and computational methods,
Novel applications in photochemistry and photobiology,
Novel interpretational approaches as well as advances in data analysis based on electronic or vibrational spectroscopy.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


