Vishikh Athavale*, , , Nikita Fedik, , , William Colglazier, , , Anders M. N. Niklasson, , , Maksim Kulichenko, , and , Sergei Tretiak,
{"title":"图形处理单元上的加速半经验激发态计算。","authors":"Vishikh Athavale*, , , Nikita Fedik, , , William Colglazier, , , Anders M. N. Niklasson, , , Maksim Kulichenko, , and , Sergei Tretiak, ","doi":"10.1021/acs.jctc.5c00980","DOIUrl":null,"url":null,"abstract":"<p >We report the development and implementation of electronic excited-state capabilities for semiempirical quantum chemical methods at both the Configuration Interaction Singles and Time-Dependent Hartree–Fock levels of theory, integrated within the PYSEQM 2.0 software package (https://github.com/lanl/PYSEQM). PYSEQM is a Python-based package designed for efficient and scalable quantum chemical simulations. Leveraging the PyTorch framework enables PYSEQM to benefit from automatic differentiation and GPU acceleration, leading to substantial performance gains in molecular property evaluations. In particular, our implementation enables efficient calculation of excited-state properties for large molecular systems. Benchmarking on systems with up to a thousand atoms demonstrates that excited-state computations can be completed in under a minute on modern GPUs, making this approach particularly suitable for high-throughput screening, real-time feedback in interactive simulations, and large-scale dynamical studies. Additionally, PYSEQM includes a machine learning interface that supports Hamiltonian parameter reoptimization and neural network training. These capabilities open new avenues for data-driven excited-state dynamics simulations, offering a path toward combining quantum chemical rigor with machine learning efficiency. Overall, this work facilitates access to excited-state quantum chemistry for large systems, while laying the foundation for future hybrid quantum-machine-learning approaches in photochemistry, photophysics, and materials discovery.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9498–9510"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"PYSEQM 2.0: Accelerated Semiempirical Excited-State Calculations on Graphical Processing Units\",\"authors\":\"Vishikh Athavale*, , , Nikita Fedik, , , William Colglazier, , , Anders M. N. Niklasson, , , Maksim Kulichenko, , and , Sergei Tretiak, \",\"doi\":\"10.1021/acs.jctc.5c00980\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We report the development and implementation of electronic excited-state capabilities for semiempirical quantum chemical methods at both the Configuration Interaction Singles and Time-Dependent Hartree–Fock levels of theory, integrated within the PYSEQM 2.0 software package (https://github.com/lanl/PYSEQM). PYSEQM is a Python-based package designed for efficient and scalable quantum chemical simulations. Leveraging the PyTorch framework enables PYSEQM to benefit from automatic differentiation and GPU acceleration, leading to substantial performance gains in molecular property evaluations. In particular, our implementation enables efficient calculation of excited-state properties for large molecular systems. Benchmarking on systems with up to a thousand atoms demonstrates that excited-state computations can be completed in under a minute on modern GPUs, making this approach particularly suitable for high-throughput screening, real-time feedback in interactive simulations, and large-scale dynamical studies. Additionally, PYSEQM includes a machine learning interface that supports Hamiltonian parameter reoptimization and neural network training. These capabilities open new avenues for data-driven excited-state dynamics simulations, offering a path toward combining quantum chemical rigor with machine learning efficiency. Overall, this work facilitates access to excited-state quantum chemistry for large systems, while laying the foundation for future hybrid quantum-machine-learning approaches in photochemistry, photophysics, and materials discovery.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"21 19\",\"pages\":\"9498–9510\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-09-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00980\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00980","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
PYSEQM 2.0: Accelerated Semiempirical Excited-State Calculations on Graphical Processing Units
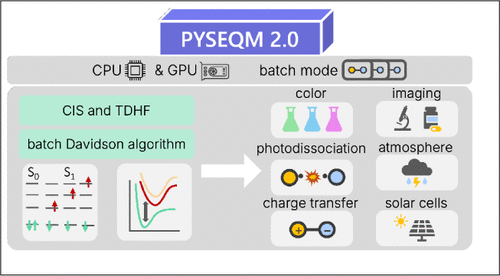
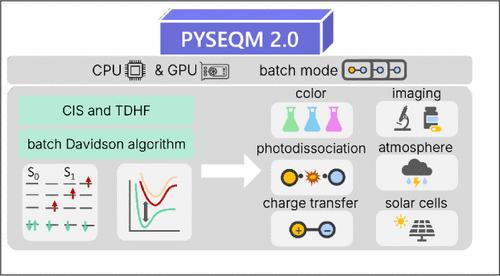
We report the development and implementation of electronic excited-state capabilities for semiempirical quantum chemical methods at both the Configuration Interaction Singles and Time-Dependent Hartree–Fock levels of theory, integrated within the PYSEQM 2.0 software package (https://github.com/lanl/PYSEQM). PYSEQM is a Python-based package designed for efficient and scalable quantum chemical simulations. Leveraging the PyTorch framework enables PYSEQM to benefit from automatic differentiation and GPU acceleration, leading to substantial performance gains in molecular property evaluations. In particular, our implementation enables efficient calculation of excited-state properties for large molecular systems. Benchmarking on systems with up to a thousand atoms demonstrates that excited-state computations can be completed in under a minute on modern GPUs, making this approach particularly suitable for high-throughput screening, real-time feedback in interactive simulations, and large-scale dynamical studies. Additionally, PYSEQM includes a machine learning interface that supports Hamiltonian parameter reoptimization and neural network training. These capabilities open new avenues for data-driven excited-state dynamics simulations, offering a path toward combining quantum chemical rigor with machine learning efficiency. Overall, this work facilitates access to excited-state quantum chemistry for large systems, while laying the foundation for future hybrid quantum-machine-learning approaches in photochemistry, photophysics, and materials discovery.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


