{"title":"具有零资历试波函数的辅助场量子蒙特卡罗方法。","authors":"Yuichiro Yoshida*, , , Luca Erhart, , , Takuma Murokoshi, , , Rika Nakagawa, , , Chihiro Mori, , , Hanae Tagami, , and , Wataru Mizukami*, ","doi":"10.1021/acs.jctc.5c00778","DOIUrl":null,"url":null,"abstract":"<p >We present an approach that uses the doubly occupied configuration interaction (DOCI) wave function as the trial wave function in phaseless auxiliary-field quantum Monte Carlo (ph-AFQMC). DOCI is a seniority-zero method focused on electron pairs. Although DOCI considers much fewer electron configurations than the complete active space (CAS) configuration interaction method, it efficiently captures the static correlation, while the consequent ph-AFQMC recovers the dynamical correlation across all orbitals. We also explore an orbital-optimized version (OO–DOCI) to further improve accuracy. We test this approach on several chemical systems, including single O–H bond breaking in water and polymer additives. In these cases, OO–DOCI-AFQMC closely matches CAS-based ph-AFQMC and even outperforms coupled-cluster singles, doubles, and perturbative triples. However, for strongly correlated systems, such as the carbon dimer and multibond dissociation in hydrogen systems and water, the method’s accuracy drops. This suggests that seniority-zero space models may be insufficient as trial wave functions in ph-AFQMC for strongly correlated systems, suggesting the need for trial wave functions in an extended space. Despite such a limitation, our study demonstrates that DOCI- and OO–DOCI-based ph-AFQMC can reduce the steep cost of CAS approaches, offering a path to accurate multireference calculations for larger, more complex systems.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9404–9414"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"The Auxiliary-Field Quantum Monte Carlo Method with Seniority-Zero Trial Wave Function\",\"authors\":\"Yuichiro Yoshida*, , , Luca Erhart, , , Takuma Murokoshi, , , Rika Nakagawa, , , Chihiro Mori, , , Hanae Tagami, , and , Wataru Mizukami*, \",\"doi\":\"10.1021/acs.jctc.5c00778\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We present an approach that uses the doubly occupied configuration interaction (DOCI) wave function as the trial wave function in phaseless auxiliary-field quantum Monte Carlo (ph-AFQMC). DOCI is a seniority-zero method focused on electron pairs. Although DOCI considers much fewer electron configurations than the complete active space (CAS) configuration interaction method, it efficiently captures the static correlation, while the consequent ph-AFQMC recovers the dynamical correlation across all orbitals. We also explore an orbital-optimized version (OO–DOCI) to further improve accuracy. We test this approach on several chemical systems, including single O–H bond breaking in water and polymer additives. In these cases, OO–DOCI-AFQMC closely matches CAS-based ph-AFQMC and even outperforms coupled-cluster singles, doubles, and perturbative triples. However, for strongly correlated systems, such as the carbon dimer and multibond dissociation in hydrogen systems and water, the method’s accuracy drops. This suggests that seniority-zero space models may be insufficient as trial wave functions in ph-AFQMC for strongly correlated systems, suggesting the need for trial wave functions in an extended space. Despite such a limitation, our study demonstrates that DOCI- and OO–DOCI-based ph-AFQMC can reduce the steep cost of CAS approaches, offering a path to accurate multireference calculations for larger, more complex systems.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"21 19\",\"pages\":\"9404–9414\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-09-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00778\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00778","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
The Auxiliary-Field Quantum Monte Carlo Method with Seniority-Zero Trial Wave Function
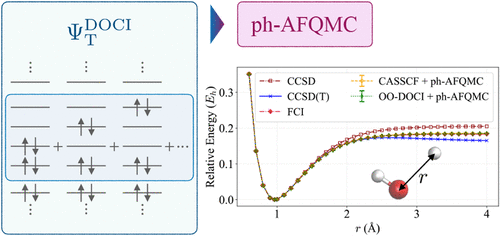
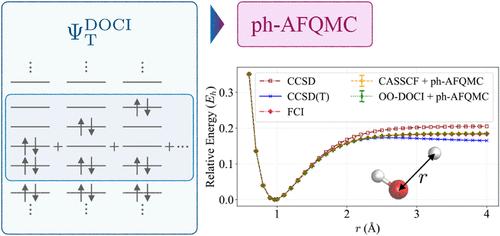
We present an approach that uses the doubly occupied configuration interaction (DOCI) wave function as the trial wave function in phaseless auxiliary-field quantum Monte Carlo (ph-AFQMC). DOCI is a seniority-zero method focused on electron pairs. Although DOCI considers much fewer electron configurations than the complete active space (CAS) configuration interaction method, it efficiently captures the static correlation, while the consequent ph-AFQMC recovers the dynamical correlation across all orbitals. We also explore an orbital-optimized version (OO–DOCI) to further improve accuracy. We test this approach on several chemical systems, including single O–H bond breaking in water and polymer additives. In these cases, OO–DOCI-AFQMC closely matches CAS-based ph-AFQMC and even outperforms coupled-cluster singles, doubles, and perturbative triples. However, for strongly correlated systems, such as the carbon dimer and multibond dissociation in hydrogen systems and water, the method’s accuracy drops. This suggests that seniority-zero space models may be insufficient as trial wave functions in ph-AFQMC for strongly correlated systems, suggesting the need for trial wave functions in an extended space. Despite such a limitation, our study demonstrates that DOCI- and OO–DOCI-based ph-AFQMC can reduce the steep cost of CAS approaches, offering a path to accurate multireference calculations for larger, more complex systems.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


