João G. F. Romeu, , , Nickolas A. Joyner, , , Elliot Kaye, , , Kirk A. Peterson*, , , Thomas T. Kawagoe, , , Samantha L. Stinson, , , Michael D. Morse*, , and , David A. Dixon*,
{"title":"单硫系锆和铪的电子结构。","authors":"João G. F. Romeu, , , Nickolas A. Joyner, , , Elliot Kaye, , , Kirk A. Peterson*, , , Thomas T. Kawagoe, , , Samantha L. Stinson, , , Michael D. Morse*, , and , David A. Dixon*, ","doi":"10.1021/acs.jpca.5c05402","DOIUrl":null,"url":null,"abstract":"<p >High-level <i>ab initio</i> CCSD(T) and spin–orbit icMRCI+Q calculations were used to predict potential energy curves (PECs) for the lowest-lying states of ZrO, ZrS, HfO, and HfS. The prediction of the ground state is basis set dependent at the icMRCI+Q level for ZrO and ZrS due to the small singlet–triplet splitting between the lowest <sup>1</sup>Σ<sup>+</sup> and <sup>3</sup>Δ states. CCSD(T) with a spin orbit correction predicted the <sup>1</sup>Σ<sup>+</sup> ground state in agreement with experiment. New all-electron basis sets were developed for Hf to improve the results over those predicted by use of effective core potentials (ECPs) that subsume the 4f electrons into the definition of the core. The use of the new DK-4f basis sets rather than ECPs became more important for HfO and HfS where there is a lack of a good core–valence separation. icMRCI+Q, CCSD(T), and DFT calculations for the spectroscopic parameters of ZrO, ZrS, HfO, and HfS were benchmarked with available experimental data. Bond dissociation energies (BDEs) of these four systems were calculated at the Feller–Peterson–Dixon (FPD) level to be 762.1 (ZrO), 543.5 (ZrS), 803.8 (HfO), and 575.1 kJ/mol (HfS), in excellent agreement with experiment. The HfS BDE was remeasured using the R3PI method, providing an updated experimental measurement of <i>D</i><sub>0</sub>(HfS) = 5.978 ± 0.002 eV = 576.8 ± 0.2 kJ/mol. This experimental value, combined with experimental measurements of the ionization energies of Hf and HfS, gives the cationic BDE of <i>D</i><sub>0</sub>(Hf<sup>+</sup>-S) = 5.124 ± 0.002 eV = 494.4 ± 0.2 kJ/mol.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"129 40","pages":"9325–9341"},"PeriodicalIF":2.8000,"publicationDate":"2025-09-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"The Electronic Structure of Zirconium and Hafnium Monochalcogenides\",\"authors\":\"João G. F. Romeu, , , Nickolas A. Joyner, , , Elliot Kaye, , , Kirk A. Peterson*, , , Thomas T. Kawagoe, , , Samantha L. Stinson, , , Michael D. Morse*, , and , David A. Dixon*, \",\"doi\":\"10.1021/acs.jpca.5c05402\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >High-level <i>ab initio</i> CCSD(T) and spin–orbit icMRCI+Q calculations were used to predict potential energy curves (PECs) for the lowest-lying states of ZrO, ZrS, HfO, and HfS. The prediction of the ground state is basis set dependent at the icMRCI+Q level for ZrO and ZrS due to the small singlet–triplet splitting between the lowest <sup>1</sup>Σ<sup>+</sup> and <sup>3</sup>Δ states. CCSD(T) with a spin orbit correction predicted the <sup>1</sup>Σ<sup>+</sup> ground state in agreement with experiment. New all-electron basis sets were developed for Hf to improve the results over those predicted by use of effective core potentials (ECPs) that subsume the 4f electrons into the definition of the core. The use of the new DK-4f basis sets rather than ECPs became more important for HfO and HfS where there is a lack of a good core–valence separation. icMRCI+Q, CCSD(T), and DFT calculations for the spectroscopic parameters of ZrO, ZrS, HfO, and HfS were benchmarked with available experimental data. Bond dissociation energies (BDEs) of these four systems were calculated at the Feller–Peterson–Dixon (FPD) level to be 762.1 (ZrO), 543.5 (ZrS), 803.8 (HfO), and 575.1 kJ/mol (HfS), in excellent agreement with experiment. The HfS BDE was remeasured using the R3PI method, providing an updated experimental measurement of <i>D</i><sub>0</sub>(HfS) = 5.978 ± 0.002 eV = 576.8 ± 0.2 kJ/mol. This experimental value, combined with experimental measurements of the ionization energies of Hf and HfS, gives the cationic BDE of <i>D</i><sub>0</sub>(Hf<sup>+</sup>-S) = 5.124 ± 0.002 eV = 494.4 ± 0.2 kJ/mol.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"129 40\",\"pages\":\"9325–9341\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-09-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.5c05402\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.5c05402","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
利用高能级从头算CCSD(T)和自旋轨道icMRCI+Q计算,预测了ZrO、ZrS、HfO和HfS最低能级的势能曲线(PECs)。基态的预测依赖于ZrO和ZrS的icMRCI+Q水平的基集,因为最低的1Σ+和3Δ态之间存在小的单重态-三重态分裂。经过自旋轨道修正的CCSD(T)预测的1Σ+基态与实验结果一致。开发了新的Hf全电子基集,以改进使用有效核电位(ECPs)预测的结果,有效核电位将4f电子包含在核的定义中。对于缺乏良好核心价分离的HfO和HfS,使用新的DK-4f基组而不是ECPs变得更加重要。用现有实验数据对ZrO、ZrS、HfO和HfS的光谱参数进行icMRCI+Q、CCSD(T)和DFT计算。在FPD (Feller-Peterson-Dixon)水平上计算了这四种体系的键离解能(BDEs)分别为762.1 (ZrO)、543.5 (ZrS)、803.8 (HfO)和575.1 kJ/mol (HfS),与实验结果吻合良好。采用R3PI法重新测定HfS的BDE,得到D0(HfS) = 5.978±0.002 eV = 576.8±0.2 kJ/mol。结合实验测量的Hf和HfS的电离能,得到D0(Hf+-S)的阳离子BDE = 5.124±0.002 eV = 494.4±0.2 kJ/mol。
The Electronic Structure of Zirconium and Hafnium Monochalcogenides
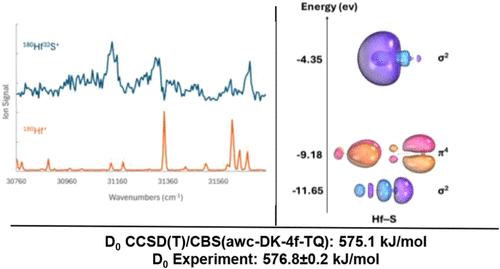
High-level ab initio CCSD(T) and spin–orbit icMRCI+Q calculations were used to predict potential energy curves (PECs) for the lowest-lying states of ZrO, ZrS, HfO, and HfS. The prediction of the ground state is basis set dependent at the icMRCI+Q level for ZrO and ZrS due to the small singlet–triplet splitting between the lowest 1Σ+ and 3Δ states. CCSD(T) with a spin orbit correction predicted the 1Σ+ ground state in agreement with experiment. New all-electron basis sets were developed for Hf to improve the results over those predicted by use of effective core potentials (ECPs) that subsume the 4f electrons into the definition of the core. The use of the new DK-4f basis sets rather than ECPs became more important for HfO and HfS where there is a lack of a good core–valence separation. icMRCI+Q, CCSD(T), and DFT calculations for the spectroscopic parameters of ZrO, ZrS, HfO, and HfS were benchmarked with available experimental data. Bond dissociation energies (BDEs) of these four systems were calculated at the Feller–Peterson–Dixon (FPD) level to be 762.1 (ZrO), 543.5 (ZrS), 803.8 (HfO), and 575.1 kJ/mol (HfS), in excellent agreement with experiment. The HfS BDE was remeasured using the R3PI method, providing an updated experimental measurement of D0(HfS) = 5.978 ± 0.002 eV = 576.8 ± 0.2 kJ/mol. This experimental value, combined with experimental measurements of the ionization energies of Hf and HfS, gives the cationic BDE of D0(Hf+-S) = 5.124 ± 0.002 eV = 494.4 ± 0.2 kJ/mol.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


