Luca Di Fiore, , , Luigi Crisci*, , , Federico Lazzari, , , Marco Mendolicchio, , and , Vincenzo Barone*,
{"title":"根据实验旋转常数验证核碱基互变异构体的基准平衡结构。","authors":"Luca Di Fiore, , , Luigi Crisci*, , , Federico Lazzari, , , Marco Mendolicchio, , and , Vincenzo Barone*, ","doi":"10.1021/acs.jpca.5c04369","DOIUrl":null,"url":null,"abstract":"<p >Tautomerism in nucleobases is a subtle yet decisive factor in key biological processes, including mutagenesis, enzymatic catalysis, and molecular recognition. Capturing the exact geometry of these fleeting forms is a demanding task, as their small energy differences are highly sensitive to stereoelectronic effects and environmental perturbations. Gas-phase high-resolution spectroscopy provides precise experimental data on individual tautomers, but interpreting these measurements in structural terms requires highly accurate quantum-chemical calculations. Here, we compute benchmark-quality structures for a representative set of canonical and noncanonical nucleobase tautomers with available experimental rotational constants. The approach combines geometry optimizations─based on CCSD(T)-F12 energies with MP2 core–valence corrections─with vibrational corrections from DFT anharmonic force fields within second-order vibrational perturbation theory. The resulting near-spectroscopic accuracy enables an unbiased comparison with experiment and demonstrates the robustness of the protocol in capturing subtle structural features. Building on this, we extend the study to fluorinated and sulfur-containing pyrimidine derivatives for which no gas-phase data are available, providing reference-quality structural parameters to guide and accelerate future high-resolution experimental and theoretical investigations.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"129 40","pages":"9217–9226"},"PeriodicalIF":2.8000,"publicationDate":"2025-09-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Benchmark Equilibrium Structures of Nucleobase Tautomers Validated Against Experimental Rotational Constants\",\"authors\":\"Luca Di Fiore, , , Luigi Crisci*, , , Federico Lazzari, , , Marco Mendolicchio, , and , Vincenzo Barone*, \",\"doi\":\"10.1021/acs.jpca.5c04369\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Tautomerism in nucleobases is a subtle yet decisive factor in key biological processes, including mutagenesis, enzymatic catalysis, and molecular recognition. Capturing the exact geometry of these fleeting forms is a demanding task, as their small energy differences are highly sensitive to stereoelectronic effects and environmental perturbations. Gas-phase high-resolution spectroscopy provides precise experimental data on individual tautomers, but interpreting these measurements in structural terms requires highly accurate quantum-chemical calculations. Here, we compute benchmark-quality structures for a representative set of canonical and noncanonical nucleobase tautomers with available experimental rotational constants. The approach combines geometry optimizations─based on CCSD(T)-F12 energies with MP2 core–valence corrections─with vibrational corrections from DFT anharmonic force fields within second-order vibrational perturbation theory. The resulting near-spectroscopic accuracy enables an unbiased comparison with experiment and demonstrates the robustness of the protocol in capturing subtle structural features. Building on this, we extend the study to fluorinated and sulfur-containing pyrimidine derivatives for which no gas-phase data are available, providing reference-quality structural parameters to guide and accelerate future high-resolution experimental and theoretical investigations.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"129 40\",\"pages\":\"9217–9226\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-09-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.5c04369\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.5c04369","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
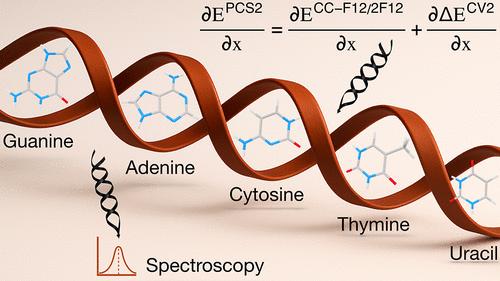
Benchmark Equilibrium Structures of Nucleobase Tautomers Validated Against Experimental Rotational Constants
Tautomerism in nucleobases is a subtle yet decisive factor in key biological processes, including mutagenesis, enzymatic catalysis, and molecular recognition. Capturing the exact geometry of these fleeting forms is a demanding task, as their small energy differences are highly sensitive to stereoelectronic effects and environmental perturbations. Gas-phase high-resolution spectroscopy provides precise experimental data on individual tautomers, but interpreting these measurements in structural terms requires highly accurate quantum-chemical calculations. Here, we compute benchmark-quality structures for a representative set of canonical and noncanonical nucleobase tautomers with available experimental rotational constants. The approach combines geometry optimizations─based on CCSD(T)-F12 energies with MP2 core–valence corrections─with vibrational corrections from DFT anharmonic force fields within second-order vibrational perturbation theory. The resulting near-spectroscopic accuracy enables an unbiased comparison with experiment and demonstrates the robustness of the protocol in capturing subtle structural features. Building on this, we extend the study to fluorinated and sulfur-containing pyrimidine derivatives for which no gas-phase data are available, providing reference-quality structural parameters to guide and accelerate future high-resolution experimental and theoretical investigations.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


