Melody Yiyuan Zhang, , , Shih-Kuang Alex Lee, , , Sharon C. Glotzer, , and , Rebecca K. Lindsey*,
{"title":"建立聚合物接枝纳米颗粒炼金术多体相互作用模型的广义机器学习框架。","authors":"Melody Yiyuan Zhang, , , Shih-Kuang Alex Lee, , , Sharon C. Glotzer, , and , Rebecca K. Lindsey*, ","doi":"10.1021/acs.jctc.5c00901","DOIUrl":null,"url":null,"abstract":"<p >Polymer-grafted nanoparticles (PGNs) serve as highly customizable building blocks for technologically relevant self-assembled nanomaterials. Physics-informed inverse design strategies are crucial for expediting exploration of the massive associated design space by identifying optimal PGN attributes, such as polymer length and grafting density, to meet a target self-assembled structure. However, their success hinges on an accurate description of how PGN interactions vary as a function of both particle positions and physical attributes. We introduce a framework to meet this need. Specifically, we develop an “alchemical” machine-learned interatomic model (ML-IAM) that describes how PGN interactions vary as a function of both inter-PGN distances <i>and</i> tunable PGN attributes, simultaneously. This model is an extension of the physics-informed and explicitly many-bodied ChIMES ML-IAM. The resulting extended ChIMES (X-ChIMES) ML-IAM is trained on potential of mean force (PMF) data for PGNs with varied polymer ligand lengths. We enable efficient training data generation by combining the forward–reverse steered molecular dynamics enhanced sampling approach with a grid-sampling scheme in the HOOMD-blue software package. We demonstrate the efficacy of ChIMES for generating coarse-grained (CG) models for PGNs with fixed design attributes, development of X-ChIMES, and its application for enhancing digital alchemy inverse-design simulations using HOOMD-blue and the ChIMES Calculator. This is the first application of ChIMES in modeling CG systems and coupling with an inverse design method to target nanomaterial self-assembly.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9853–9867"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"A Generalized Machine-Learning Framework for Developing Alchemical Many-Body Interaction Models for Polymer-Grafted Nanoparticles\",\"authors\":\"Melody Yiyuan Zhang, , , Shih-Kuang Alex Lee, , , Sharon C. Glotzer, , and , Rebecca K. Lindsey*, \",\"doi\":\"10.1021/acs.jctc.5c00901\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Polymer-grafted nanoparticles (PGNs) serve as highly customizable building blocks for technologically relevant self-assembled nanomaterials. Physics-informed inverse design strategies are crucial for expediting exploration of the massive associated design space by identifying optimal PGN attributes, such as polymer length and grafting density, to meet a target self-assembled structure. However, their success hinges on an accurate description of how PGN interactions vary as a function of both particle positions and physical attributes. We introduce a framework to meet this need. Specifically, we develop an “alchemical” machine-learned interatomic model (ML-IAM) that describes how PGN interactions vary as a function of both inter-PGN distances <i>and</i> tunable PGN attributes, simultaneously. This model is an extension of the physics-informed and explicitly many-bodied ChIMES ML-IAM. The resulting extended ChIMES (X-ChIMES) ML-IAM is trained on potential of mean force (PMF) data for PGNs with varied polymer ligand lengths. We enable efficient training data generation by combining the forward–reverse steered molecular dynamics enhanced sampling approach with a grid-sampling scheme in the HOOMD-blue software package. We demonstrate the efficacy of ChIMES for generating coarse-grained (CG) models for PGNs with fixed design attributes, development of X-ChIMES, and its application for enhancing digital alchemy inverse-design simulations using HOOMD-blue and the ChIMES Calculator. This is the first application of ChIMES in modeling CG systems and coupling with an inverse design method to target nanomaterial self-assembly.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"21 19\",\"pages\":\"9853–9867\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-09-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00901\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00901","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
A Generalized Machine-Learning Framework for Developing Alchemical Many-Body Interaction Models for Polymer-Grafted Nanoparticles
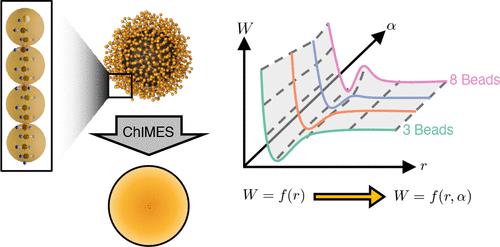
Polymer-grafted nanoparticles (PGNs) serve as highly customizable building blocks for technologically relevant self-assembled nanomaterials. Physics-informed inverse design strategies are crucial for expediting exploration of the massive associated design space by identifying optimal PGN attributes, such as polymer length and grafting density, to meet a target self-assembled structure. However, their success hinges on an accurate description of how PGN interactions vary as a function of both particle positions and physical attributes. We introduce a framework to meet this need. Specifically, we develop an “alchemical” machine-learned interatomic model (ML-IAM) that describes how PGN interactions vary as a function of both inter-PGN distances and tunable PGN attributes, simultaneously. This model is an extension of the physics-informed and explicitly many-bodied ChIMES ML-IAM. The resulting extended ChIMES (X-ChIMES) ML-IAM is trained on potential of mean force (PMF) data for PGNs with varied polymer ligand lengths. We enable efficient training data generation by combining the forward–reverse steered molecular dynamics enhanced sampling approach with a grid-sampling scheme in the HOOMD-blue software package. We demonstrate the efficacy of ChIMES for generating coarse-grained (CG) models for PGNs with fixed design attributes, development of X-ChIMES, and its application for enhancing digital alchemy inverse-design simulations using HOOMD-blue and the ChIMES Calculator. This is the first application of ChIMES in modeling CG systems and coupling with an inverse design method to target nanomaterial self-assembly.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


