{"title":"对眼咽肌营养不良异质性的认识。","authors":"Kyriaki Kekou, Constantinos Papadopoulos, Maria Svingou, Margarita Chrysanthou-Piterou, Evangelia Nitsa, Danai Veltra, Nikos Marinakis, Faidon-Nikolaos Tilemis, Parissis Dimitrios, Marianthi Arnaoutoglou, Maria Moschou, Sophia Xirou, Christos Bakirtzis, Georgios Tsivgoulis, Giorgos-Konstantinos Papadimas, Christalena Sofocleous","doi":"10.1007/s10048-025-00849-0","DOIUrl":null,"url":null,"abstract":"<p><p>Oculopharyngeal muscular dystrophy (OPMD) is a rare, adult-onset, autosomal dominant myopathy characterized by variability in the age of onset and disease progression. However, its pathogenesis and phenotypic variability remain poorly understood. The disorder is caused by an expansion of a short polyalanine tract in the poly(A) binding protein nuclear 1 (PABPN1) gene. This study presents data from 23 patients across 19 Greek families with pathogenic PABPN1 expansions, including demographic and laboratory data, as well as molecular and electron microscopy findings. Eight distinct trinucleotide expansion genotypes were identified. Electron microscopy consistently demonstrated mitochondrial abnormalities, including swelling, disrupted cristae and atypical lipid inclusions. Clinical heterogeneity was observed at both inter- and intrafamilial levels, and milder phenotypes were generally linked to smaller alleles. Notably, maternally inherited expansions were associated with an earlier disease onset and more severe progression in affected offspring. Given the genetic variability observed in the cohort, the presence of a founder effect could not be supported. A significant degree of underdiagnosis or diagnostic delay was noted, largely attributable to the rarity and clinical heterogeneity of the disease. The observed intrafamilial heterogeneity - particularly in maternally inherited expansions - supports previous reports suggesting that mitochondrial dysfunction may contribute to transgenerational disease progression in the context of a dominant, causative nuclear variant.</p>","PeriodicalId":56106,"journal":{"name":"Neurogenetics","volume":"26 1","pages":"68"},"PeriodicalIF":1.2000,"publicationDate":"2025-09-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12460444/pdf/","citationCount":"0","resultStr":"{\"title\":\"Insights into the heterogeneity of oculopharyngeal muscular dystrophy.\",\"authors\":\"Kyriaki Kekou, Constantinos Papadopoulos, Maria Svingou, Margarita Chrysanthou-Piterou, Evangelia Nitsa, Danai Veltra, Nikos Marinakis, Faidon-Nikolaos Tilemis, Parissis Dimitrios, Marianthi Arnaoutoglou, Maria Moschou, Sophia Xirou, Christos Bakirtzis, Georgios Tsivgoulis, Giorgos-Konstantinos Papadimas, Christalena Sofocleous\",\"doi\":\"10.1007/s10048-025-00849-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Oculopharyngeal muscular dystrophy (OPMD) is a rare, adult-onset, autosomal dominant myopathy characterized by variability in the age of onset and disease progression. However, its pathogenesis and phenotypic variability remain poorly understood. The disorder is caused by an expansion of a short polyalanine tract in the poly(A) binding protein nuclear 1 (PABPN1) gene. This study presents data from 23 patients across 19 Greek families with pathogenic PABPN1 expansions, including demographic and laboratory data, as well as molecular and electron microscopy findings. Eight distinct trinucleotide expansion genotypes were identified. Electron microscopy consistently demonstrated mitochondrial abnormalities, including swelling, disrupted cristae and atypical lipid inclusions. Clinical heterogeneity was observed at both inter- and intrafamilial levels, and milder phenotypes were generally linked to smaller alleles. Notably, maternally inherited expansions were associated with an earlier disease onset and more severe progression in affected offspring. Given the genetic variability observed in the cohort, the presence of a founder effect could not be supported. A significant degree of underdiagnosis or diagnostic delay was noted, largely attributable to the rarity and clinical heterogeneity of the disease. The observed intrafamilial heterogeneity - particularly in maternally inherited expansions - supports previous reports suggesting that mitochondrial dysfunction may contribute to transgenerational disease progression in the context of a dominant, causative nuclear variant.</p>\",\"PeriodicalId\":56106,\"journal\":{\"name\":\"Neurogenetics\",\"volume\":\"26 1\",\"pages\":\"68\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2025-09-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12460444/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Neurogenetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s10048-025-00849-0\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Neurogenetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10048-025-00849-0","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Insights into the heterogeneity of oculopharyngeal muscular dystrophy.
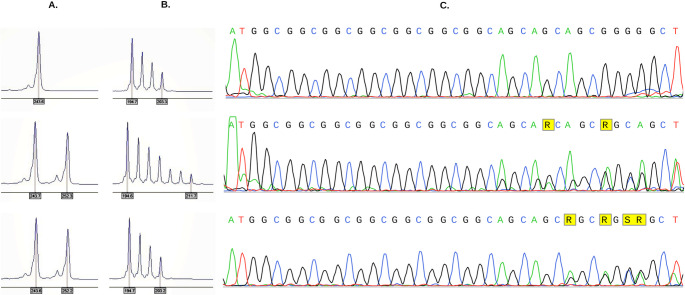
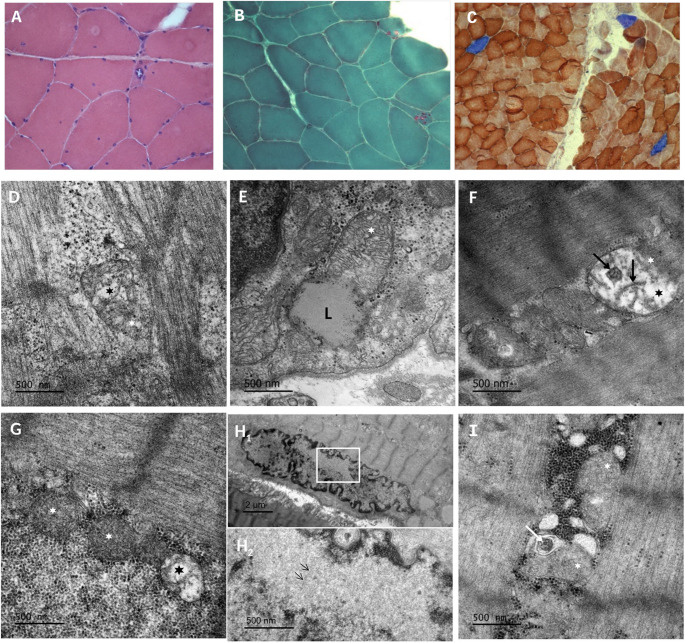
Oculopharyngeal muscular dystrophy (OPMD) is a rare, adult-onset, autosomal dominant myopathy characterized by variability in the age of onset and disease progression. However, its pathogenesis and phenotypic variability remain poorly understood. The disorder is caused by an expansion of a short polyalanine tract in the poly(A) binding protein nuclear 1 (PABPN1) gene. This study presents data from 23 patients across 19 Greek families with pathogenic PABPN1 expansions, including demographic and laboratory data, as well as molecular and electron microscopy findings. Eight distinct trinucleotide expansion genotypes were identified. Electron microscopy consistently demonstrated mitochondrial abnormalities, including swelling, disrupted cristae and atypical lipid inclusions. Clinical heterogeneity was observed at both inter- and intrafamilial levels, and milder phenotypes were generally linked to smaller alleles. Notably, maternally inherited expansions were associated with an earlier disease onset and more severe progression in affected offspring. Given the genetic variability observed in the cohort, the presence of a founder effect could not be supported. A significant degree of underdiagnosis or diagnostic delay was noted, largely attributable to the rarity and clinical heterogeneity of the disease. The observed intrafamilial heterogeneity - particularly in maternally inherited expansions - supports previous reports suggesting that mitochondrial dysfunction may contribute to transgenerational disease progression in the context of a dominant, causative nuclear variant.
期刊介绍:
Neurogenetics publishes findings that contribute to a better understanding of the genetic basis of normal and abnormal function of the nervous system. Neurogenetic disorders are the main focus of the journal. Neurogenetics therefore includes findings in humans and other organisms that help understand neurological disease mechanisms and publishes papers from many different fields such as biophysics, cell biology, human genetics, neuroanatomy, neurochemistry, neurology, neuropathology, neurosurgery and psychiatry.
All papers submitted to Neurogenetics should be of sufficient immediate importance to justify urgent publication. They should present new scientific results. Data merely confirming previously published findings are not acceptable.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


