{"title":"利用下一代测序技术鉴定21-羟化酶缺乏症CYP21A2变异的优化同源序列比对","authors":"Yibo Chen, Qi Yu, Lisha Ge, Lixin Weng, Xiaoli Pan, Xiaoxia Zhou, Nani Zhou, Yanjie Wang, Jia Jia, Haibo Li","doi":"10.2147/RMHP.S514355","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>This study aimed to develop a novel homologous sequence analysis technique using high-throughput sequencing data to enhance CYP21A2 mutation detection. The approach leverages next-generation sequencing to overcome existing limitations and improve 21-hydroxylase deficiency diagnostic accuracy.</p><p><strong>Methods: </strong>From April 21, 2022, to February 21, 2023, a total of 100 unrelated participants were enrolled at the Women and Children's Hospital of Ningbo University, selected based on clinical manifestations and genetic testing results. The study used next-generation sequencing combined with a homologous sequence alignment (HSA) algorithm, which calculated the sequencing read ratios from homologous regions to identify pathogenic or likely pathogenic variants in the CYP21A2 gene. All detected variants were further validated using long-range PCR or multiplex ligation-dependent probe amplification. The accuracy of the HSA algorithm was systematically assessed.</p><p><strong>Results: </strong>Among the 100 participants, 84 were identified as carriers of CYP21A2 mutations, while 16 were diagnosed with 21-hydroxylase deficiency. A total of 107 pathogenic mutations were detected using the homologous sequence alignment algorithm, comprising of 99 single nucleotide variants or insertions/deletions, 6 copy number variants, and 8 fusion mutations. Additionally, eight cases of CYP21A2-CYP21A1P gene conversions were identified based on HSA scores and confirmed through long-range PCR or multiplex ligation-dependent probe amplification. The algorithm demonstrated a positive predictive value of 96.26% for identifying mutations in CYP21A2. The most frequently observed mutations included c.955C > T, c.844G > T, c.293-13C > G, c.518T > A, and exon-level deletions.</p><p><strong>Conclusion: </strong>In genetic testing, particularly when addressing misalignment challenges associated with highly homologous genes such as CYP21A2, application of the HSA algorithm enables accurate mutation detection using commonly employed short-read sequencing methods. Through the characterization of homologous sequence features and optimization of the HSA algorithm, accurate mutation detection can be achieved in more homologous gene families (eg, HBA1/HBA2, SMN1/SMN2, GBA/GBAP1).</p>","PeriodicalId":56009,"journal":{"name":"Risk Management and Healthcare Policy","volume":"18 ","pages":"3063-3078"},"PeriodicalIF":2.0000,"publicationDate":"2025-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12449869/pdf/","citationCount":"0","resultStr":"{\"title\":\"Optimized Homologous Sequence Alignment for the Identification of CYP21A2 Variants in 21-Hydroxylase Deficiency Using Next-Generation Sequencing Technology.\",\"authors\":\"Yibo Chen, Qi Yu, Lisha Ge, Lixin Weng, Xiaoli Pan, Xiaoxia Zhou, Nani Zhou, Yanjie Wang, Jia Jia, Haibo Li\",\"doi\":\"10.2147/RMHP.S514355\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objective: </strong>This study aimed to develop a novel homologous sequence analysis technique using high-throughput sequencing data to enhance CYP21A2 mutation detection. The approach leverages next-generation sequencing to overcome existing limitations and improve 21-hydroxylase deficiency diagnostic accuracy.</p><p><strong>Methods: </strong>From April 21, 2022, to February 21, 2023, a total of 100 unrelated participants were enrolled at the Women and Children's Hospital of Ningbo University, selected based on clinical manifestations and genetic testing results. The study used next-generation sequencing combined with a homologous sequence alignment (HSA) algorithm, which calculated the sequencing read ratios from homologous regions to identify pathogenic or likely pathogenic variants in the CYP21A2 gene. All detected variants were further validated using long-range PCR or multiplex ligation-dependent probe amplification. The accuracy of the HSA algorithm was systematically assessed.</p><p><strong>Results: </strong>Among the 100 participants, 84 were identified as carriers of CYP21A2 mutations, while 16 were diagnosed with 21-hydroxylase deficiency. A total of 107 pathogenic mutations were detected using the homologous sequence alignment algorithm, comprising of 99 single nucleotide variants or insertions/deletions, 6 copy number variants, and 8 fusion mutations. Additionally, eight cases of CYP21A2-CYP21A1P gene conversions were identified based on HSA scores and confirmed through long-range PCR or multiplex ligation-dependent probe amplification. The algorithm demonstrated a positive predictive value of 96.26% for identifying mutations in CYP21A2. The most frequently observed mutations included c.955C > T, c.844G > T, c.293-13C > G, c.518T > A, and exon-level deletions.</p><p><strong>Conclusion: </strong>In genetic testing, particularly when addressing misalignment challenges associated with highly homologous genes such as CYP21A2, application of the HSA algorithm enables accurate mutation detection using commonly employed short-read sequencing methods. Through the characterization of homologous sequence features and optimization of the HSA algorithm, accurate mutation detection can be achieved in more homologous gene families (eg, HBA1/HBA2, SMN1/SMN2, GBA/GBAP1).</p>\",\"PeriodicalId\":56009,\"journal\":{\"name\":\"Risk Management and Healthcare Policy\",\"volume\":\"18 \",\"pages\":\"3063-3078\"},\"PeriodicalIF\":2.0000,\"publicationDate\":\"2025-09-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12449869/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Risk Management and Healthcare Policy\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.2147/RMHP.S514355\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"HEALTH CARE SCIENCES & SERVICES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Risk Management and Healthcare Policy","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.2147/RMHP.S514355","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"HEALTH CARE SCIENCES & SERVICES","Score":null,"Total":0}
Optimized Homologous Sequence Alignment for the Identification of CYP21A2 Variants in 21-Hydroxylase Deficiency Using Next-Generation Sequencing Technology.
Objective: This study aimed to develop a novel homologous sequence analysis technique using high-throughput sequencing data to enhance CYP21A2 mutation detection. The approach leverages next-generation sequencing to overcome existing limitations and improve 21-hydroxylase deficiency diagnostic accuracy.
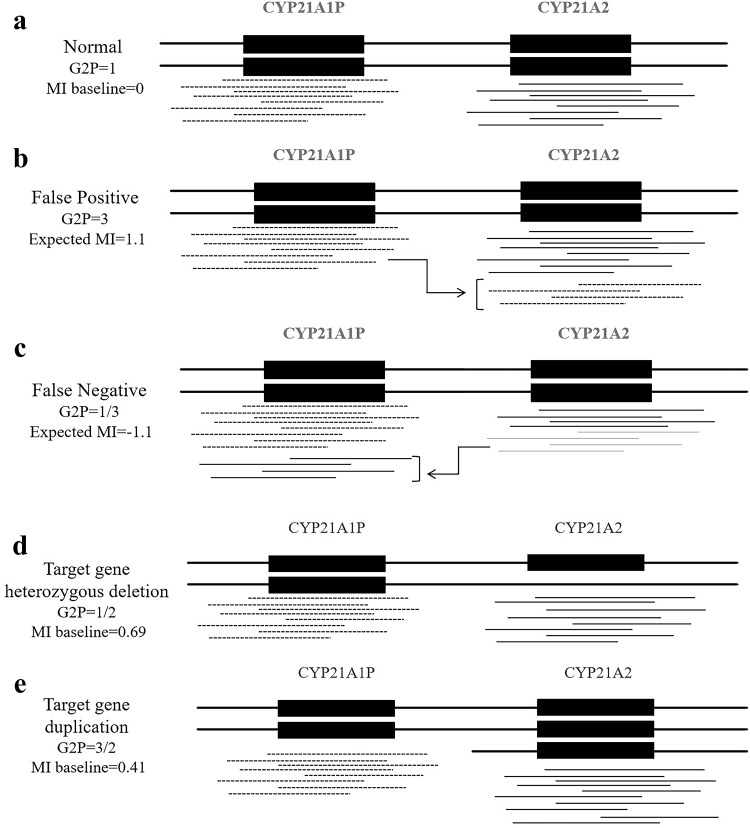
Methods: From April 21, 2022, to February 21, 2023, a total of 100 unrelated participants were enrolled at the Women and Children's Hospital of Ningbo University, selected based on clinical manifestations and genetic testing results. The study used next-generation sequencing combined with a homologous sequence alignment (HSA) algorithm, which calculated the sequencing read ratios from homologous regions to identify pathogenic or likely pathogenic variants in the CYP21A2 gene. All detected variants were further validated using long-range PCR or multiplex ligation-dependent probe amplification. The accuracy of the HSA algorithm was systematically assessed.
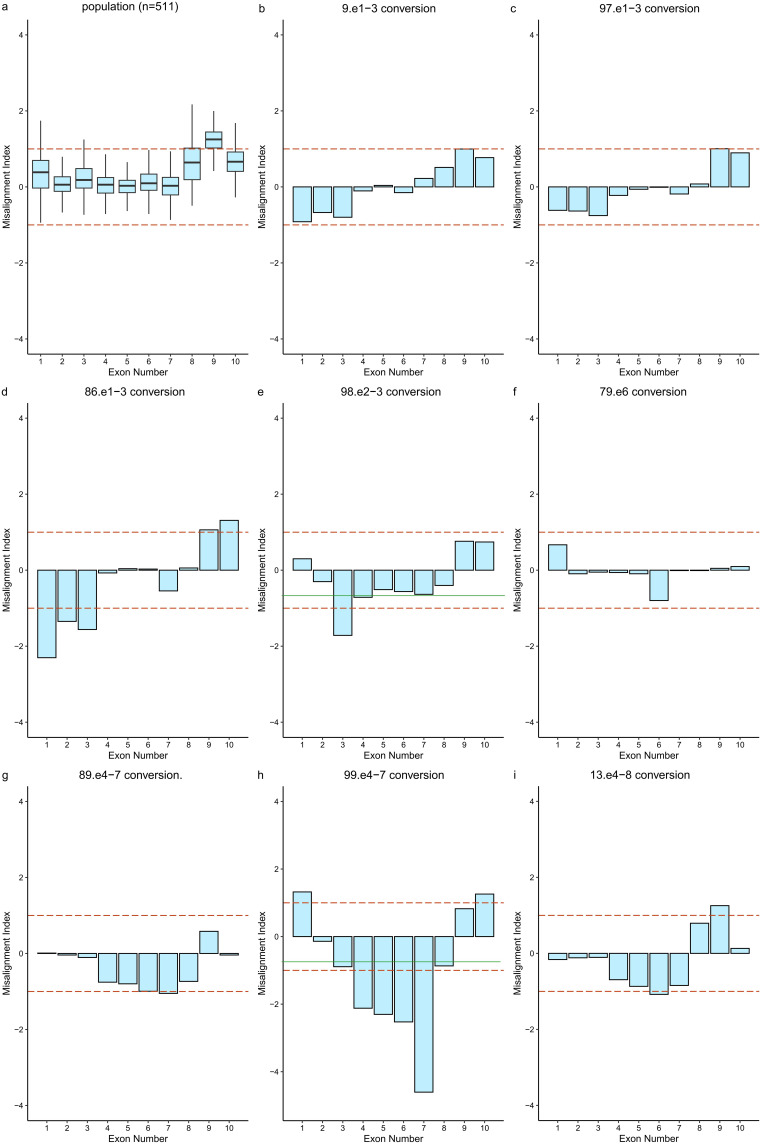
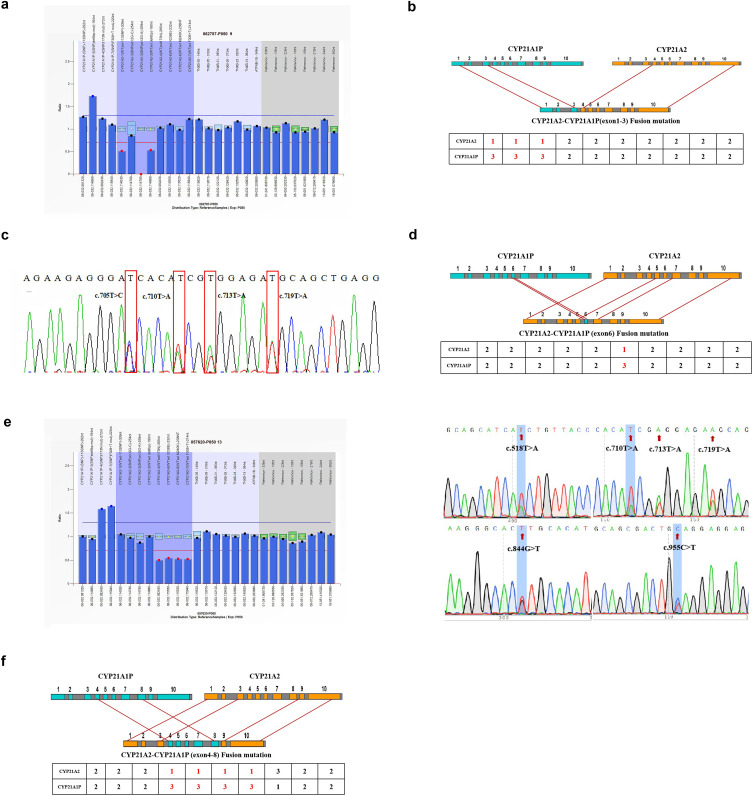
Results: Among the 100 participants, 84 were identified as carriers of CYP21A2 mutations, while 16 were diagnosed with 21-hydroxylase deficiency. A total of 107 pathogenic mutations were detected using the homologous sequence alignment algorithm, comprising of 99 single nucleotide variants or insertions/deletions, 6 copy number variants, and 8 fusion mutations. Additionally, eight cases of CYP21A2-CYP21A1P gene conversions were identified based on HSA scores and confirmed through long-range PCR or multiplex ligation-dependent probe amplification. The algorithm demonstrated a positive predictive value of 96.26% for identifying mutations in CYP21A2. The most frequently observed mutations included c.955C > T, c.844G > T, c.293-13C > G, c.518T > A, and exon-level deletions.
Conclusion: In genetic testing, particularly when addressing misalignment challenges associated with highly homologous genes such as CYP21A2, application of the HSA algorithm enables accurate mutation detection using commonly employed short-read sequencing methods. Through the characterization of homologous sequence features and optimization of the HSA algorithm, accurate mutation detection can be achieved in more homologous gene families (eg, HBA1/HBA2, SMN1/SMN2, GBA/GBAP1).
期刊介绍:
Risk Management and Healthcare Policy is an international, peer-reviewed, open access journal focusing on all aspects of public health, policy and preventative measures to promote good health and improve morbidity and mortality in the population. Specific topics covered in the journal include:
Public and community health
Policy and law
Preventative and predictive healthcare
Risk and hazard management
Epidemiology, detection and screening
Lifestyle and diet modification
Vaccination and disease transmission/modification programs
Health and safety and occupational health
Healthcare services provision
Health literacy and education
Advertising and promotion of health issues
Health economic evaluations and resource management
Risk Management and Healthcare Policy focuses on human interventional and observational research. The journal welcomes submitted papers covering original research, clinical and epidemiological studies, reviews and evaluations, guidelines, expert opinion and commentary, and extended reports. Case reports will only be considered if they make a valuable and original contribution to the literature. The journal does not accept study protocols, animal-based or cell line-based studies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


