{"title":"通过计算建模和仿真大规模鉴定PARP7抑制剂","authors":"Xiaochen Yang, Baolin Liu and Daixi Li","doi":"10.1039/D5NJ02053K","DOIUrl":null,"url":null,"abstract":"<p >Poly(ADP-ribose) polymerases (PARPs) are attractive therapeutic targets for cancer. This study focuses on PARP7, a mono(ADP-ribose) polymerase with emerging potential in cancer therapy due to its roles in immune response and tumorigenesis. Using computational modeling, we screened millions of compounds through molecular docking, machine learning based on molecular fingerprints, molecular dynamics (MD) simulations, and ADME profiling. We identified promising PARP7 inhibitor candidates exhibiting higher binding affinity than NAD<small><sup>+</sup></small> and comparable affinity to RBN2397, with favorable binding energies and pharmacological properties. MD simulations confirmed complex stability, while interaction analysis revealed key binding residues including conserved residues (Y564/H532) and hydrophobic residues (F575/I542). <em>In silico</em> ADME predictions indicated favorable drug-like properties and pharmacokinetic profiles. This work establishes a foundation for developing novel PARP7 inhibitors, offering new therapeutic strategies for cancer.</p>","PeriodicalId":95,"journal":{"name":"New Journal of Chemistry","volume":" 37","pages":" 16320-16332"},"PeriodicalIF":2.5000,"publicationDate":"2025-08-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Large-scale identification of PARP7 inhibitors via computational modeling and simulation\",\"authors\":\"Xiaochen Yang, Baolin Liu and Daixi Li\",\"doi\":\"10.1039/D5NJ02053K\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Poly(ADP-ribose) polymerases (PARPs) are attractive therapeutic targets for cancer. This study focuses on PARP7, a mono(ADP-ribose) polymerase with emerging potential in cancer therapy due to its roles in immune response and tumorigenesis. Using computational modeling, we screened millions of compounds through molecular docking, machine learning based on molecular fingerprints, molecular dynamics (MD) simulations, and ADME profiling. We identified promising PARP7 inhibitor candidates exhibiting higher binding affinity than NAD<small><sup>+</sup></small> and comparable affinity to RBN2397, with favorable binding energies and pharmacological properties. MD simulations confirmed complex stability, while interaction analysis revealed key binding residues including conserved residues (Y564/H532) and hydrophobic residues (F575/I542). <em>In silico</em> ADME predictions indicated favorable drug-like properties and pharmacokinetic profiles. This work establishes a foundation for developing novel PARP7 inhibitors, offering new therapeutic strategies for cancer.</p>\",\"PeriodicalId\":95,\"journal\":{\"name\":\"New Journal of Chemistry\",\"volume\":\" 37\",\"pages\":\" 16320-16332\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2025-08-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"New Journal of Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/nj/d5nj02053k\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"New Journal of Chemistry","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/nj/d5nj02053k","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Large-scale identification of PARP7 inhibitors via computational modeling and simulation
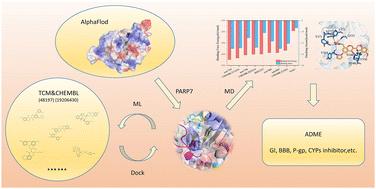
Poly(ADP-ribose) polymerases (PARPs) are attractive therapeutic targets for cancer. This study focuses on PARP7, a mono(ADP-ribose) polymerase with emerging potential in cancer therapy due to its roles in immune response and tumorigenesis. Using computational modeling, we screened millions of compounds through molecular docking, machine learning based on molecular fingerprints, molecular dynamics (MD) simulations, and ADME profiling. We identified promising PARP7 inhibitor candidates exhibiting higher binding affinity than NAD+ and comparable affinity to RBN2397, with favorable binding energies and pharmacological properties. MD simulations confirmed complex stability, while interaction analysis revealed key binding residues including conserved residues (Y564/H532) and hydrophobic residues (F575/I542). In silico ADME predictions indicated favorable drug-like properties and pharmacokinetic profiles. This work establishes a foundation for developing novel PARP7 inhibitors, offering new therapeutic strategies for cancer.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


