光氧化还原/镍双催化C(sp2)-C(sp3)偶联反应中镍-碳配合物Ni-C键解离焓的DFT研究
IF 2.2
3区 化学
Q2 CHEMISTRY, ORGANIC
引用次数: 0
摘要
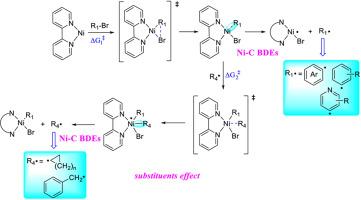
传统的过渡金属催化可以有效地形成C(sp2)-C(sp2)键,但由于C(sp3)氧化加成和金属化缓慢,以及β-氢化物的消除倾向,C(sp2)-C(sp3)偶联仍然具有挑战性。一个显著的进步是双催化平台的发展,它协同整合了过渡金属催化剂和光氧化还原催化剂,为构建具有挑战性的C(sp2)-C(sp3)键提供了强有力的策略。镍配合物因其成本效益、丰富的性质和催化效率而被认可,已成为该领域的关键参与者。它们与光氧化还原催化的相容性使得镍中间体与(杂)芳基卤化物或烷基自由基配合,在反应周期中形成关键的Ni-C键的独特机制途径成为可能。通过均溶键解离焓(BDE)测量准确量化这些Ni-C键的强度对于理解催化行为至关重要。在这项研究中,我们使用十种密度泛函理论(DFT)方法系统地评估了14种镍碳配合物,并根据实验数据对计算的BDE值进行了基准测试。M06功能优于其他功能,表现出最低的均方根误差(RMSE)为2.7 kcal/mol。利用这一发现,在Ni/光氧化还原双催化条件下,M06功能函数被应用于模型配合物I和II中Ni - c BDEs的评价,同时探测取代基效应。通过自然键轨道(NBO)分析和前沿分子轨道能量评估,进一步了解Ni-C BDE的变化规律。过渡态表征进一步揭示了Ni-C键强度与活化自由能之间的直接关系,建立了将热力学稳定性与机制可行性联系起来的结构-活性框架。本文章由计算机程序翻译,如有差异,请以英文原文为准。
DFT study on Ni–C bond dissociation enthalpies of nickel-carbon complexes in photoredox/nickel dual catalysis C(sp2)-C(sp3) coupling reactions
Traditional transition metal catalysis efficiently forms C(sp2)-C(sp2) bonds, but C(sp2)-C(sp3) couplings remain challenging due to sluggish C(sp3) oxidative addition and metalation, and the propensity for β-hydride elimination. A notable advancement is the development of dual catalytic platforms that synergistically integrate transition metal catalysts with photoredox catalysts, offering a powerful strategy for constructing challenging C(sp2)-C(sp3) linkages. Nickel complexes, recognized for their cost-effectiveness, earth-abundant nature, and catalytic efficiency, have emerged as pivotal players in this field. Their compatibility with photoredox catalysis enables a unique mechanistic pathway where nickel intermediates coordinate with (hetero)aryl halides or alkyl radicals, forming critical Ni–C bonds during the reaction cycle. Accurately quantifying the strength of these Ni–C bonds through homolytic bond dissociation enthalpy (BDE) measurements hold critical importance for understanding catalytic behavior. In this study, we systematically evaluated 14 nickel-carbon complexes using ten density functional theory (DFT) methodologies, benchmarking calculated BDE values against experimental data. The M06 functional outperformed others, exhibiting the lowest root-mean-square error (RMSE) of 2.7 kcal/mol. Leveraging this finding, the M06 functional was applied to evaluate Ni–C BDEs in model complexes I and II under Ni/photoredox dual catalytic conditions, while probing substituent effects. Supplementary investigations, natural bond orbital (NBO) analyses and frontier molecular orbital energy assessments, were conducted to further understand the variation patterns of Ni–C BDE. Transition state characterization further uncovered a direct correlation between Ni–C bond strength and activation free energies, establishing a structure-activity framework that links thermodynamic stability with mechanistic feasibility.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Tetrahedron
化学-有机化学
CiteScore
3.90
自引率
4.80%
发文量
439
审稿时长
34 days
期刊介绍:
Tetrahedron publishes full accounts of research having outstanding significance in the broad field of organic chemistry and its related disciplines, such as organic materials and bio-organic chemistry.
Regular papers in Tetrahedron are expected to represent detailed accounts of an original study having substantially greater scope and details than that found in a communication, as published in Tetrahedron Letters.
Tetrahedron also publishes thematic collections of papers as special issues and ''Reports'', commissioned in-depth reviews providing a comprehensive overview of a research area.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


