利用邻接矩阵幂和原子序数序列预测分子性质
IF 4.3
2区 工程技术
Q2 ENGINEERING, CHEMICAL
引用次数: 0
摘要
物理化学性质如沸点的准确预测是核心的虚拟筛选,工艺设计,并在化学信息学监管评估。在这里,我们引入了一个由加权邻接矩阵幂衍生的排列不变分子描述符,其中边权编码键类型(single=1, double=2, triple=3, aromatic=1.5)和一个填充到固定长度的有序原子序数序列。该描述符捕获基于行走的拓扑信息,同时为多达134个原子的分子保持紧凑、固定大小的表示。我们使用支持向量回归、随机森林、极端梯度增强(XGBoost)和定向消息传递神经网络(通过Chemprop的D - MPNN)对五种已建立的表示(MACCS密钥、摩根指纹、莫德雷德描述符、库伦矩阵、Weisfeiler-Lehman图核)的性能进行了基准测试。在包含5,432种小有机化合物沸点的实验数据集上,我们的描述符与XGBoost配对实现了5倍交叉验证,平均绝对误差为18.52±0.34°C,均方根误差为27.16±0.32°C, R2=0.898±0.002。虽然高维Mordred描述符产生了最准确的预测(MAE=10.54±0.23°C, R2=0.960±0.003),但我们的描述符提供了简单指纹和更复杂的基于图的表示的平衡替代方案。该研究表明,计算效率高,基于行走的描述符可以在沸点回归任务中实现鲁棒和可解释的性能。它的低维性和跨模型的可泛化性使其成为更广泛的热物性预测和集成到图神经网络体系结构中的有前途的工具。本文章由计算机程序翻译,如有差异,请以英文原文为准。
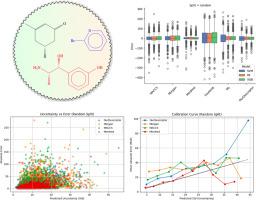
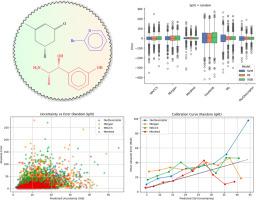
Predicting molecular properties using adjacency matrix powers and atomic number sequences
Accurate prediction of physicochemical properties such as boiling points is central to virtual screening, process design, and regulatory assessment in cheminformatics. Here, we introduce a permutation-invariant molecular descriptor derived from powers of a weighted adjacency matrix-where edge weights encode bond types (single=1, double=2, triple=3, aromatic=1.5)-and a sorted atomic number sequence padded to a fixed length. The descriptor captures walk-based topological information while maintaining a compact, fixed-size representation for molecules up to 134 atoms. We benchmark its performance against five established representations (MACCS keys, Morgan fingerprints, Mordred descriptors, Coulomb matrix, Weisfeiler-Lehman graph kernels) using Support Vector Regression, Random Forest, Extreme Gradient Boosting (XGBoost), and a Directed Message-Passing Neural Network (D-MPNN via Chemprop).
On a diverse dataset of 5432 small organic compounds with experimentally measured boiling points, our descriptor paired with XGBoost achieved a 5-fold cross-validation mean absolute error of 18.52±0.34°C, root mean square error of 27.16±0.32°C, and =0.898±0.002. While high-dimensional Mordred descriptors yielded the most accurate predictions (MAE=10.54±0.23°C, =0.960±0.003), our descriptor presents a balanced alternative to simple fingerprints and more complex graph-based representations.
This study demonstrates that a computationally efficient, walk-based descriptor can achieve robust and interpretable performance in boiling-point regression tasks. Its low dimensionality and generalizability across models make it a promising tool for broader thermophysical property prediction and integration into graph-neural-network architectures.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Chemical Engineering Science
工程技术-工程:化工
CiteScore
7.50
自引率
8.50%
发文量
1025
审稿时长
50 days
期刊介绍:
Chemical engineering enables the transformation of natural resources and energy into useful products for society. It draws on and applies natural sciences, mathematics and economics, and has developed fundamental engineering science that underpins the discipline.
Chemical Engineering Science (CES) has been publishing papers on the fundamentals of chemical engineering since 1951. CES is the platform where the most significant advances in the discipline have ever since been published. Chemical Engineering Science has accompanied and sustained chemical engineering through its development into the vibrant and broad scientific discipline it is today.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


