Pablo Iruzubieta, David Pellerin, Catherine Ashton, Felipe Villa, Mathilde Renaud, Marie-Josée Dicaire, Matt C Danzi, Mayra Aldecoa, Jean Mathieu, Rami Massie, Colin H Chalk, Anne-Louise Lafontaine, François Evoy, Marie-France Rioux, Jean-Denis Brisson, Kym M Boycott, Henry Houlden, Matthis Synofzik, Roberta La Piana, Stephan Zuchner, Antoine Duquette, Bernard Brais
{"title":"描述SCA27B的致病阈值和表型谱:来自一个大型法裔加拿大队列的发现。","authors":"Pablo Iruzubieta, David Pellerin, Catherine Ashton, Felipe Villa, Mathilde Renaud, Marie-Josée Dicaire, Matt C Danzi, Mayra Aldecoa, Jean Mathieu, Rami Massie, Colin H Chalk, Anne-Louise Lafontaine, François Evoy, Marie-France Rioux, Jean-Denis Brisson, Kym M Boycott, Henry Houlden, Matthis Synofzik, Roberta La Piana, Stephan Zuchner, Antoine Duquette, Bernard Brais","doi":"10.1007/s00415-025-13387-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Autosomal dominant spinocerebellar ataxia 27B (SCA27B), caused by an intronic (GAA•TTC) repeat expansion in FGF14, is a common cause of late-onset cerebellar ataxia, but its genotypic and phenotypic spectrum remains to be fully established.</p><p><strong>Methods: </strong>We analysed the FGF14 (GAA•TTC) repeat expansion in a cohort of 134 patients with ataxia and 822 controls from Quebec. We conducted segregation study in large families to further characterize intergenerational repeat instability.</p><p><strong>Results: </strong>We found a significant enrichment of (GAA•TTC)<sub>≥200</sub> alleles in the ataxia cohort compared to controls (53.0%, 71/134, vs 3.6%, 30/822, p < 0.0001), including for (GAA•TTC)<sub>200-249</sub> alleles (8.2% vs 2.6%, p = 0.0026). We identified 12 ataxic patients with a phenotype compatible with SCA27B carrying a (GAA•TTC)<sub>200-249</sub> expansion supporting the pathogenicity of these alleles in some patients. We further delineated the phenotype of 125 symptomatic individuals from 69 families who carried an FGF14 (GAA•TTC)<sub>≥200</sub> repeat expansion. Patients with (GAA•TTC)<sub>200-249</sub>, (GAA•TTC)<sub>250-299</sub>, and (GAA•TTC)<sub>≥300</sub> had a similar phenotype. We observed that 14% of patients with episodic symptoms (13/92) had severe episodes that were initially misdiagnosed as stroke, vestibular neuritis, Wernicke's encephalopathy, or seizures.</p><p><strong>Discussion and conclusion: </strong>This large cohort demonstrates that (GAA•TTC)<sub>200-249</sub> alleles are enriched in patients with ataxia compared to controls and can be pathogenic for SCA27B, supporting the need to define a lower pathogenic threshold in the presence of specific clinical criteria.</p>","PeriodicalId":16558,"journal":{"name":"Journal of Neurology","volume":"272 9","pages":"636"},"PeriodicalIF":4.6000,"publicationDate":"2025-09-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12450228/pdf/","citationCount":"0","resultStr":"{\"title\":\"Delineating the pathogenic threshold and phenotypic spectrum of SCA27B: findings from a large French-Canadian cohort.\",\"authors\":\"Pablo Iruzubieta, David Pellerin, Catherine Ashton, Felipe Villa, Mathilde Renaud, Marie-Josée Dicaire, Matt C Danzi, Mayra Aldecoa, Jean Mathieu, Rami Massie, Colin H Chalk, Anne-Louise Lafontaine, François Evoy, Marie-France Rioux, Jean-Denis Brisson, Kym M Boycott, Henry Houlden, Matthis Synofzik, Roberta La Piana, Stephan Zuchner, Antoine Duquette, Bernard Brais\",\"doi\":\"10.1007/s00415-025-13387-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Autosomal dominant spinocerebellar ataxia 27B (SCA27B), caused by an intronic (GAA•TTC) repeat expansion in FGF14, is a common cause of late-onset cerebellar ataxia, but its genotypic and phenotypic spectrum remains to be fully established.</p><p><strong>Methods: </strong>We analysed the FGF14 (GAA•TTC) repeat expansion in a cohort of 134 patients with ataxia and 822 controls from Quebec. We conducted segregation study in large families to further characterize intergenerational repeat instability.</p><p><strong>Results: </strong>We found a significant enrichment of (GAA•TTC)<sub>≥200</sub> alleles in the ataxia cohort compared to controls (53.0%, 71/134, vs 3.6%, 30/822, p < 0.0001), including for (GAA•TTC)<sub>200-249</sub> alleles (8.2% vs 2.6%, p = 0.0026). We identified 12 ataxic patients with a phenotype compatible with SCA27B carrying a (GAA•TTC)<sub>200-249</sub> expansion supporting the pathogenicity of these alleles in some patients. We further delineated the phenotype of 125 symptomatic individuals from 69 families who carried an FGF14 (GAA•TTC)<sub>≥200</sub> repeat expansion. Patients with (GAA•TTC)<sub>200-249</sub>, (GAA•TTC)<sub>250-299</sub>, and (GAA•TTC)<sub>≥300</sub> had a similar phenotype. We observed that 14% of patients with episodic symptoms (13/92) had severe episodes that were initially misdiagnosed as stroke, vestibular neuritis, Wernicke's encephalopathy, or seizures.</p><p><strong>Discussion and conclusion: </strong>This large cohort demonstrates that (GAA•TTC)<sub>200-249</sub> alleles are enriched in patients with ataxia compared to controls and can be pathogenic for SCA27B, supporting the need to define a lower pathogenic threshold in the presence of specific clinical criteria.</p>\",\"PeriodicalId\":16558,\"journal\":{\"name\":\"Journal of Neurology\",\"volume\":\"272 9\",\"pages\":\"636\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2025-09-20\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12450228/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Neurology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s00415-025-13387-4\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Neurology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s00415-025-13387-4","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Delineating the pathogenic threshold and phenotypic spectrum of SCA27B: findings from a large French-Canadian cohort.
Background: Autosomal dominant spinocerebellar ataxia 27B (SCA27B), caused by an intronic (GAA•TTC) repeat expansion in FGF14, is a common cause of late-onset cerebellar ataxia, but its genotypic and phenotypic spectrum remains to be fully established.
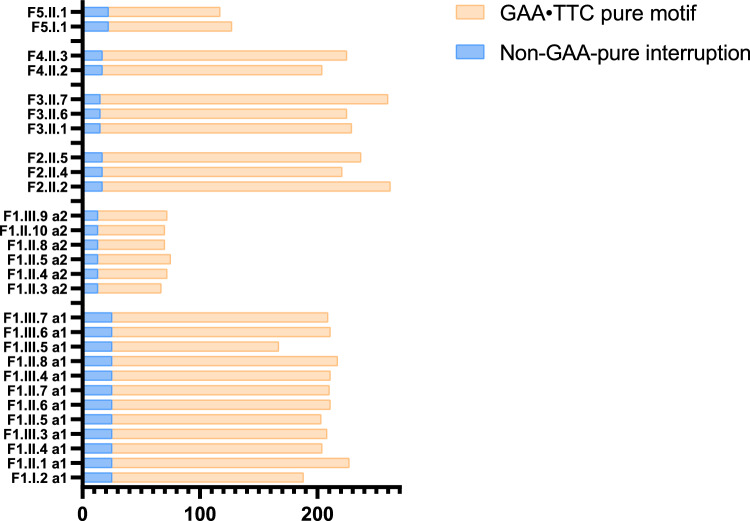
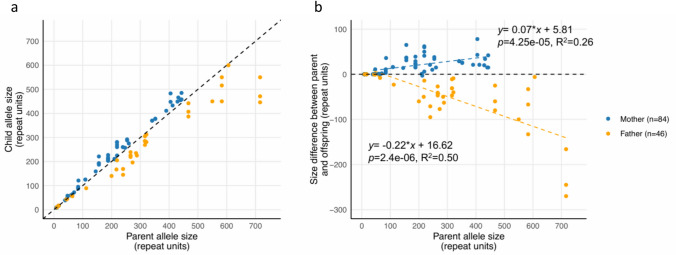
Methods: We analysed the FGF14 (GAA•TTC) repeat expansion in a cohort of 134 patients with ataxia and 822 controls from Quebec. We conducted segregation study in large families to further characterize intergenerational repeat instability.
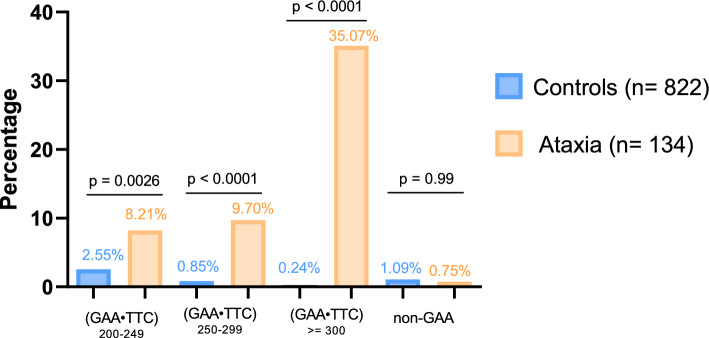
Results: We found a significant enrichment of (GAA•TTC)≥200 alleles in the ataxia cohort compared to controls (53.0%, 71/134, vs 3.6%, 30/822, p < 0.0001), including for (GAA•TTC)200-249 alleles (8.2% vs 2.6%, p = 0.0026). We identified 12 ataxic patients with a phenotype compatible with SCA27B carrying a (GAA•TTC)200-249 expansion supporting the pathogenicity of these alleles in some patients. We further delineated the phenotype of 125 symptomatic individuals from 69 families who carried an FGF14 (GAA•TTC)≥200 repeat expansion. Patients with (GAA•TTC)200-249, (GAA•TTC)250-299, and (GAA•TTC)≥300 had a similar phenotype. We observed that 14% of patients with episodic symptoms (13/92) had severe episodes that were initially misdiagnosed as stroke, vestibular neuritis, Wernicke's encephalopathy, or seizures.
Discussion and conclusion: This large cohort demonstrates that (GAA•TTC)200-249 alleles are enriched in patients with ataxia compared to controls and can be pathogenic for SCA27B, supporting the need to define a lower pathogenic threshold in the presence of specific clinical criteria.
期刊介绍:
The Journal of Neurology is an international peer-reviewed journal which provides a source for publishing original communications and reviews on clinical neurology covering the whole field.
In addition, Letters to the Editors serve as a forum for clinical cases and the exchange of ideas which highlight important new findings. A section on Neurological progress serves to summarise the major findings in certain fields of neurology. Commentaries on new developments in clinical neuroscience, which may be commissioned or submitted, are published as editorials.
Every neurologist interested in the current diagnosis and treatment of neurological disorders needs access to the information contained in this valuable journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


